
SUMMARY: The American Cancer Society estimates that in 2014, over 46,000 people will be diagnosed with Pancreatic Cancer in the United States and close to 40,000 people will die of the disease. Some important risk factors for Pancreatic Cancer include increasing age, obesity, smoking history, genetic predisposition, exposure to certain dyes and chemicals, heavy alcohol use and pancreatitis. The best chance for long term survival is complete surgical resection, although this may not be feasible in a majority of the patients, as they present with advanced disease at the time of diagnosis. Based on the National Cancer Data Base, the 5 year observed survival rate for patients diagnosed with exocrine cancer of the pancreas is 14% for those with Stage IA disease and 1% for those with Stage IV disease. The FDA recently granted Breakthrough Therapy Designation status for the combination treatment that consists of two vaccines, GVAX and CRS-207, for patients with advanced Pancreatic Carcinoma. This designation was based on the following study. A phase II clinical trial was conducted in which the authors took a novel approach and tested a combination of two vaccines in patients with metastatic pancreatic adenocarcinoma.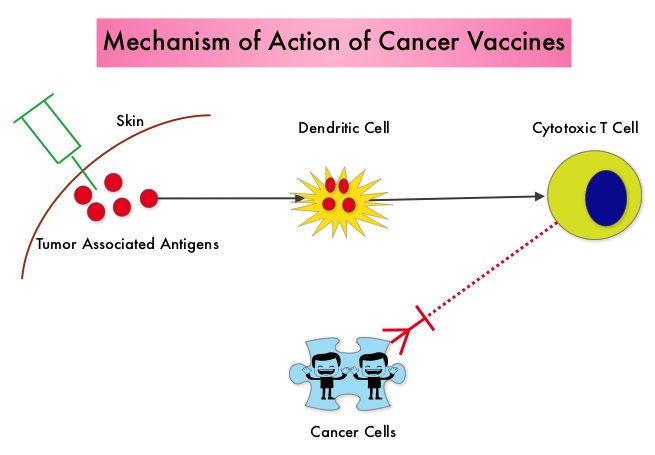 Traditional vaccination against specific bacterial and viral infections involves the injection of the specific weakened bacteria/virus or a structural component of the bacteria or virus. The body then mounts an immune response and is ready to respond to an infection associated with that specific bacteria or virus. Use of vaccines in cancer treatment is based on the same principle. The two vaccines studied were GVAX and CRS-207. GVAX is an allogeneic whole cell vaccine developed from pancreatic cancer cell lines. These cancer cells are irradiated, to prevent them from dividing and are genetically modified to secrete GM-CSF (Granulocyte Macrophage Colony Stimulating Factor). GM-CSF is important for the growth and activation of dendritic cells also known as Antigen Presenting Cells. This vaccine when injected attracts the dendritic cells to the vaccine injection site and the dendritic cells in turn, pick up the antigens from the vaccine and present them to the patient’s immune system. The immune system then mounts a response by activating tumor specific T-cells. This vaccine therefore theoretically boosts the body’s immune system to fight the patient’s tumor, without causing collateral damage. The second vaccine CRS-207 is live-attenuated (weakened) Listeria monocytogenes bacterium which expresses mesothelin and stimulates innate and adaptive immunity. It is genetically engineered to elicit an immune response against the tumor-associated antigen mesothelin, which has been shown to be expressed at higher levels on pancreatic cancer cells than on normal cells. Previous studies have demonstrated that survival can be improved by induction of mesothelin specific T-cell responses. In this study, 90 patients with metastatic pancreatic adenocarcinoma were randomly assigned in a 2:1 ratio to receive two doses of GVAX followed by four doses of CRS-207 or six doses of GVAX alone. Treatment was given every 3 weeks and low-dose CYTOXAN® (Cyclophosphamide) was given IV, the day before GVAX in both groups, to inhibit regulatory (suppressive) T-cells. More than 80% of the patients had at least one prior treatment for metastatic disease and 50% had two or more prior treatments. The primary endpoint was overall survival. Secondary endpoints included safety, clinical and immune responses. At a planned interim analysis, the median overall survival was 6.1 months with the combination of two vaccines vs 3.9 months with GVAX alone (HR=0.54, P=0.011), a 46% reduction in risk of death with the combination immunotherapy. The median overall survival in patients who received three total doses which included at least two doses of GVAX and at least one dose of CRS-207 was 9.7 months compared to 4.6 months for GVAX alone (HR=0.44, P=0.0074), a 56% reduction in the risk of death. In the subgroup of patients who had two or more prior chemotherapy regimens, combination immunotherapy given as third line therapy or greater resulted in a median overall survival of 5.1 months vs 3.7 months with GVAX alone (HR=0.34, P=0.001), a 66% reduction in risk of death. Stabilization of tumor marker CA19-9, was seen in 32% of patients receiving combination immunotherapy vs 13% in those who received GVAX alone (P=0.06). The one year survival probability doubled with the dual vaccine with an estimated one year survival of 24% for the combination immunotherapy group and 12% for the GVAX alone group. Toxicities included local reactions after GVAX and transient fevers, chills, and lymphopenia after CRS-207 administration. The authors concluded that immunotherapy with a combination of two vaccines improved overall survival in patients with metastatic pancreatic carcinoma, who had failed prior therapies. Le DT, Wang-Gillam A, Picozzi V, et al. J Clin Oncol 32, 2014 (suppl 3; abstr 177)
Traditional vaccination against specific bacterial and viral infections involves the injection of the specific weakened bacteria/virus or a structural component of the bacteria or virus. The body then mounts an immune response and is ready to respond to an infection associated with that specific bacteria or virus. Use of vaccines in cancer treatment is based on the same principle. The two vaccines studied were GVAX and CRS-207. GVAX is an allogeneic whole cell vaccine developed from pancreatic cancer cell lines. These cancer cells are irradiated, to prevent them from dividing and are genetically modified to secrete GM-CSF (Granulocyte Macrophage Colony Stimulating Factor). GM-CSF is important for the growth and activation of dendritic cells also known as Antigen Presenting Cells. This vaccine when injected attracts the dendritic cells to the vaccine injection site and the dendritic cells in turn, pick up the antigens from the vaccine and present them to the patient’s immune system. The immune system then mounts a response by activating tumor specific T-cells. This vaccine therefore theoretically boosts the body’s immune system to fight the patient’s tumor, without causing collateral damage. The second vaccine CRS-207 is live-attenuated (weakened) Listeria monocytogenes bacterium which expresses mesothelin and stimulates innate and adaptive immunity. It is genetically engineered to elicit an immune response against the tumor-associated antigen mesothelin, which has been shown to be expressed at higher levels on pancreatic cancer cells than on normal cells. Previous studies have demonstrated that survival can be improved by induction of mesothelin specific T-cell responses. In this study, 90 patients with metastatic pancreatic adenocarcinoma were randomly assigned in a 2:1 ratio to receive two doses of GVAX followed by four doses of CRS-207 or six doses of GVAX alone. Treatment was given every 3 weeks and low-dose CYTOXAN® (Cyclophosphamide) was given IV, the day before GVAX in both groups, to inhibit regulatory (suppressive) T-cells. More than 80% of the patients had at least one prior treatment for metastatic disease and 50% had two or more prior treatments. The primary endpoint was overall survival. Secondary endpoints included safety, clinical and immune responses. At a planned interim analysis, the median overall survival was 6.1 months with the combination of two vaccines vs 3.9 months with GVAX alone (HR=0.54, P=0.011), a 46% reduction in risk of death with the combination immunotherapy. The median overall survival in patients who received three total doses which included at least two doses of GVAX and at least one dose of CRS-207 was 9.7 months compared to 4.6 months for GVAX alone (HR=0.44, P=0.0074), a 56% reduction in the risk of death. In the subgroup of patients who had two or more prior chemotherapy regimens, combination immunotherapy given as third line therapy or greater resulted in a median overall survival of 5.1 months vs 3.7 months with GVAX alone (HR=0.34, P=0.001), a 66% reduction in risk of death. Stabilization of tumor marker CA19-9, was seen in 32% of patients receiving combination immunotherapy vs 13% in those who received GVAX alone (P=0.06). The one year survival probability doubled with the dual vaccine with an estimated one year survival of 24% for the combination immunotherapy group and 12% for the GVAX alone group. Toxicities included local reactions after GVAX and transient fevers, chills, and lymphopenia after CRS-207 administration. The authors concluded that immunotherapy with a combination of two vaccines improved overall survival in patients with metastatic pancreatic carcinoma, who had failed prior therapies. Le DT, Wang-Gillam A, Picozzi V, et al. J Clin Oncol 32, 2014 (suppl 3; abstr 177)
Month: September 2014
Circulating Tumor Cells and Response to Chemotherapy in Metastatic Breast Cancer SWOG S0500
SUMMARY: Circulating tumor cells (CTCs) are epithelial cells that are shed into the circulation from a primary or metastatic tumor. After being shed, CTCs can remain in the circulation or undergo apoptosis. Evaluation of CTCs during the course of disease and treatment has prognostic value. Because of the very low concentrations of CTCs (1 CTC in the background of millions of normal hematopoietic cells) in the peripheral blood, different technologies have been developed that will allow enrichment and detection of these CTCs.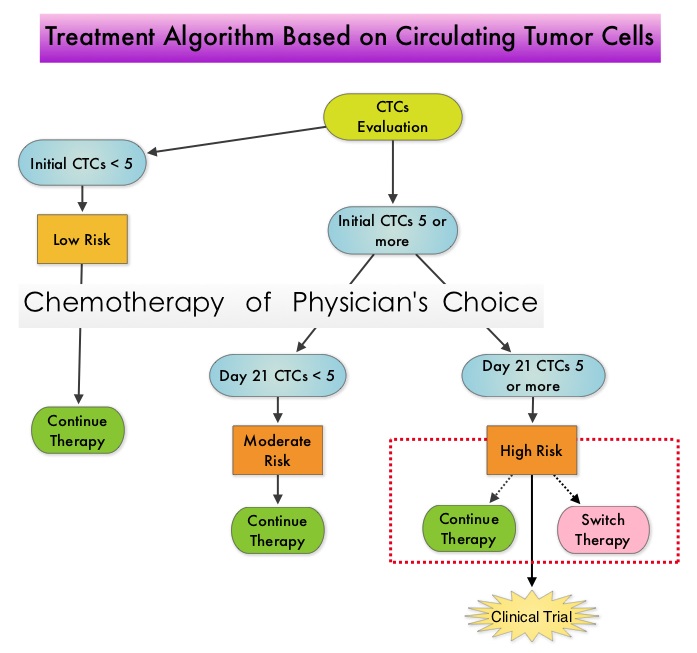 One such technology is the CellSearch® system which is the first FDA-approved test for CTC assessment, in the peripheral blood of Metastatic Breast Cancer (MBC) patients. This automated system is able to enrich the peripheral blood sample with CTCs and the cells then are fluorescently stained for CytoKeratins (CK8,18 and 19), Common Leukocyte Antigen (CD45) and a nuclear dye (DAPI). CTCs are identified when they are CK and DAPI positive and CD45 negative. In essence, CTC assessment, is a real time, peripheral blood evaluation (“Liquid Biopsy”) in MBC patients. Previously published studies have concluded that in patients with MBC, increased levels of CTCs prior to administration of a new therapy was associated with poor outcomes and failure of CTCs to drop to below 5 CTCs per 7.5 mL of peripheral blood at 3- 5 weeks after systemic therapy initiation, predicted worse Progression Free Survival (PFS) and Overall Survival (OS), compared to those who did not have increased CTCs at baseline or had increased CTCs at baseline and but not at 3-5 weeks after therapy. With this background, a randomized study was conducted to assess whether changing treatment after one cycle of first line chemotherapy, in those with a persistent increase in CTCs, improved OS. Evaluable patients were initially divided into two groups – Group A (N=276) included patients who did not have increased CTCs at baseline and Group B (N=288) included patients who had 5 or more CTCs per 7.5 mL of peripheral blood. Eligible patients were chemotherapy naïve for MBC and were treated with single agent chemotherapy. The choice of chemotherapy was at the discretion of the attending physician. Patients in Group A remained on initial therapy until disease progression whereas patients in Group B had CTC evaluation at Day 22 and those with decreased CTCs remained on the initial therapy (N=165). Patients who had persistently increased CTCs at Day 22 (N=123), were then randomly assigned to either continue the initial therapy (Group C1) or switch to a different chemotherapy regimen (Group C2). The median Overall Survival for Groups A, B, and C (C1 and C2 combined) were 35 months, 23 months, and 13 months, respectively (P <0.001). There was no difference in median Overall Survival between Groups C1 and C2 (10.7 and 12.5 months, respectively (P = 0.98). The authors concluded that CTCs in patients with Metastatic Breast Cancer receiving first line chemotherapy has significant prognostic value and changing to a different chemotherapy regimen based on persistently increased CTCs after 3 weeks of first line chemotherapy, had no impact in prolonging Overall Survival. This group of patients (C1 and C2) should be encouraged to enroll in clinical trials as standard chemotherapy may not be as effective. CTC count can prognosticate Progression Free Survival and Overall Survival early in the treatment course thereby allowing customized care. Further, CTC enumeration, unlike mucin based serum biomarkers such as CEA and CA15-3, better correlates with clinical and pathological characteristics of the disease. Smerage JB, Barlow WE, Hortobagyi GN, et al. DOI: 10.1200/JCO.2014.56.2561
One such technology is the CellSearch® system which is the first FDA-approved test for CTC assessment, in the peripheral blood of Metastatic Breast Cancer (MBC) patients. This automated system is able to enrich the peripheral blood sample with CTCs and the cells then are fluorescently stained for CytoKeratins (CK8,18 and 19), Common Leukocyte Antigen (CD45) and a nuclear dye (DAPI). CTCs are identified when they are CK and DAPI positive and CD45 negative. In essence, CTC assessment, is a real time, peripheral blood evaluation (“Liquid Biopsy”) in MBC patients. Previously published studies have concluded that in patients with MBC, increased levels of CTCs prior to administration of a new therapy was associated with poor outcomes and failure of CTCs to drop to below 5 CTCs per 7.5 mL of peripheral blood at 3- 5 weeks after systemic therapy initiation, predicted worse Progression Free Survival (PFS) and Overall Survival (OS), compared to those who did not have increased CTCs at baseline or had increased CTCs at baseline and but not at 3-5 weeks after therapy. With this background, a randomized study was conducted to assess whether changing treatment after one cycle of first line chemotherapy, in those with a persistent increase in CTCs, improved OS. Evaluable patients were initially divided into two groups – Group A (N=276) included patients who did not have increased CTCs at baseline and Group B (N=288) included patients who had 5 or more CTCs per 7.5 mL of peripheral blood. Eligible patients were chemotherapy naïve for MBC and were treated with single agent chemotherapy. The choice of chemotherapy was at the discretion of the attending physician. Patients in Group A remained on initial therapy until disease progression whereas patients in Group B had CTC evaluation at Day 22 and those with decreased CTCs remained on the initial therapy (N=165). Patients who had persistently increased CTCs at Day 22 (N=123), were then randomly assigned to either continue the initial therapy (Group C1) or switch to a different chemotherapy regimen (Group C2). The median Overall Survival for Groups A, B, and C (C1 and C2 combined) were 35 months, 23 months, and 13 months, respectively (P <0.001). There was no difference in median Overall Survival between Groups C1 and C2 (10.7 and 12.5 months, respectively (P = 0.98). The authors concluded that CTCs in patients with Metastatic Breast Cancer receiving first line chemotherapy has significant prognostic value and changing to a different chemotherapy regimen based on persistently increased CTCs after 3 weeks of first line chemotherapy, had no impact in prolonging Overall Survival. This group of patients (C1 and C2) should be encouraged to enroll in clinical trials as standard chemotherapy may not be as effective. CTC count can prognosticate Progression Free Survival and Overall Survival early in the treatment course thereby allowing customized care. Further, CTC enumeration, unlike mucin based serum biomarkers such as CEA and CA15-3, better correlates with clinical and pathological characteristics of the disease. Smerage JB, Barlow WE, Hortobagyi GN, et al. DOI: 10.1200/JCO.2014.56.2561
Improved Survival with Bevacizumab in Advanced Cervical Cancer
SUMMARY: Cervical cancer is the fourth most common cancer affecting women, worldwide. It is also the fourth most common cause of cancer death. With the availability of widespread screening techniques and HPV vaccination in the U.S., the incidence of cervical cancer is declining. Treatment of advanced cervical cancer continues to be a challenge. The FDA recently approved AVASTIN® (Bevacizumab) for the treatment of persistent, recurrent or metastatic cervical cancer, in combination with TAXOL® (Paclitaxel) and Cisplatin or TAXOL® and HYCAMTIN® (Topotecan). The approval was based on the results of an international, randomized, four-arm, 2×2 factorial design trial with two primary comparisons of Overall Survival (OS). The first comparison was between AVASTIN® plus chemotherapy versus chemotherapy alone. The second comparison of OS was between the platinum doublet versus the non-platinum doublet chemotherapy irrespective of addition of AVASTIN®. AVASTIN®, a humanized Vascular Endothelial Growth Factor (VEGF) targeted monoclonal antibody, has demonstrated single-agent activity in heavily pretreated patients with recurrent cervical carcinoma in phase II trials. In this randomized study, 452 enrolled patients with metastatic, persistent or recurrent cervical cancer received one of the four treatment regimens using a 2×2 factorial design. The four treatment groups included a) Cisplatin 50 mg/m2 plus TAXOL® (Paclitaxel) 135 or 175 mg/m2 (N=114), b) HYCAMTIN® (Topotecan) 0.75 mg/m2 given on D1 thru D3 plus TAXOL® 175 mg/m2 given on Day 1 (N=111), c) Cisplatin 50 mg/m2 plus TAXOL® 135 or 175 mg/m2 given along with AVASTIN® 15mg/kg on Day 1 (N=115) and d) HYCAMTIN® 0.75 mg/m2 given on D1 thru D3 plus TAXOL® 175 mg/m2 given on Day 1, along with AVASTIN® 15mg/kg on day 1 (N=112). Treatment was given every 21 days until disease progression, the development of unacceptable toxicities or a complete response was noted. The primary end point was Overall Survival and secondary endpoints included Progression Free Survival (PFS) and Response Rate (RR). When outcomes were analyzed, HYCAMTIN® based chemotherapy was not superior to Cisplatin based chemotherapy, regardless of prior exposure to Cisplatin. At a median follow-up of 20.8 months, the addition of AVASTIN® to chemotherapy resulted in a significantly longer median Overall Survival (17 vs 13.3 months; HR=0.71; P=0.004), significantly longer median PFS (8.2 vs 5.9 months; HR=0.67; P=0.002) and Response Rate (48% vs 36%; P=0.008), compared to combination chemotherapy alone. With regards to the second primary comparison of Overall Survival, the TAXOL® plus HYCAMTIN® with or without AVASTIN® groups did not demonstrate an improvement in Overall Survival compared to the TAXOL® plus Cisplatin with or without AVASTIN® groups. The benefit with added AVASTIN® was noted in all subgroups regardless of age, race, performance status and prior platinum exposure. Treatment was in general well tolerated without significant reduction in quality of life. As was seen in other tumor types, AVASTIN® based chemotherapy regimen was associated with a higher incidence of hypertension and thromboembolic events. The authors concluded that the addition of AVASTIN® to combination chemotherapy significantly decreased the risk of death in patients with recurrent, persistent, or metastatic cervical cancer. It can also be concluded from this study that the TAXOL® with HYCAMTIN® plus AVASTIN® is an acceptable alternative for women with advanced cervical cancer, who are not candidates for Cisplatin based chemotherapy. Tewari KS, Sill MW, Long HJ, et al. N Engl J Med 2014; 370:734-743
Confirmatory open-label, single-arm, multicenter phase 2 study of the BiTE antibody, Blinatumomab in patients (pts) with relapsed/refractory B-precursor acute lymphoblastic leukemia (r/r ALL)
SUMMARY:The FDA in July 2014, granted Breakthrough Therapy Designation to Blinatumomab, a bispecific T cell engager (BiTE) antibody, for adults with Philadelphia-negative (Ph-) Relapsed/Refractory B-precursor Acute Lymphoblastic Leukemia (ALL). BiTE® technology engages the body’s immune system to detect and target malignant cells. These modified antibodies are designed to engage two different targets simultaneously, thereby placing the T cells within reach of the targeted cancer cell and facilitating apoptosis of the cancer cell. BiTE antibodies are currently being investigated to treat a wide variety of malignancies. Blinatumomab is an investigational BiTE® antibody designed to direct the patients T cells against CD19, a protein found on the surface of B-cell derived leukemias and lymphomas.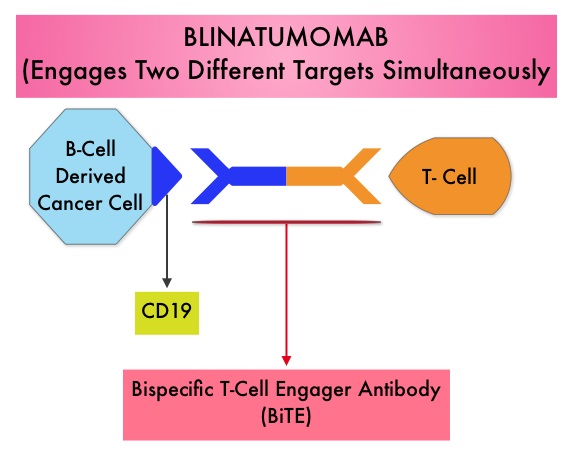 The Breakthrough Therapy Designation to Blinatumomab was based on a Phase II study in which 189 patients with Philadelphia chromosome negative ALL were enrolled. The median age was 39 years, and patients had their 1st relapse and were refractory to post hematopoietic stem cell transplantation less than 12 months before. About a third of the patients had at least 2 salvage therapies. Blinatumomab was given by continuous IV infusion, 4 weeks on and 2 weeks off for up to 5 cycles and the median number of cycles given were 2. The primary endpoint was complete remission (CR) or CR with partial hematological recovery (CRh) within the first 2 cycles of treatment. At the time of primary analysis, 43% of patients achieved a CR or CRh and 80% of responses occurred within cycle 1. Further, the Complete Remissions (CR) and CR with partial hematological recovery (CRh) were seen in all subgroups of patients, although this was more pronounced in those with less than 50% bone marrow blasts. The median Relapse Free Survival and Overall survival were 5.9 months and 6.1 months respectively. The most frequent grade 3 adverse events were febrile neutropenia, neutropenia, and anemia, occurring in 26%, 15%, and 15% of patients, respectively. The authors concluded that Blinatumomab has significant single agent antileukemia activity in a difficult-to-treat population with Relapsed and Refractory Acute Lymphoblastic Leukemia. Clinical trials will hopefully address whether Blinatumomab can serve as a bridge to transplantation, in patients with Relapsed and Refractory B-cell ALL. Topp MS, Goekbuget N, Stein AS, et al. J Clin Oncol 32:5s, 2014 (suppl; abstr 7005)</s
The Breakthrough Therapy Designation to Blinatumomab was based on a Phase II study in which 189 patients with Philadelphia chromosome negative ALL were enrolled. The median age was 39 years, and patients had their 1st relapse and were refractory to post hematopoietic stem cell transplantation less than 12 months before. About a third of the patients had at least 2 salvage therapies. Blinatumomab was given by continuous IV infusion, 4 weeks on and 2 weeks off for up to 5 cycles and the median number of cycles given were 2. The primary endpoint was complete remission (CR) or CR with partial hematological recovery (CRh) within the first 2 cycles of treatment. At the time of primary analysis, 43% of patients achieved a CR or CRh and 80% of responses occurred within cycle 1. Further, the Complete Remissions (CR) and CR with partial hematological recovery (CRh) were seen in all subgroups of patients, although this was more pronounced in those with less than 50% bone marrow blasts. The median Relapse Free Survival and Overall survival were 5.9 months and 6.1 months respectively. The most frequent grade 3 adverse events were febrile neutropenia, neutropenia, and anemia, occurring in 26%, 15%, and 15% of patients, respectively. The authors concluded that Blinatumomab has significant single agent antileukemia activity in a difficult-to-treat population with Relapsed and Refractory Acute Lymphoblastic Leukemia. Clinical trials will hopefully address whether Blinatumomab can serve as a bridge to transplantation, in patients with Relapsed and Refractory B-cell ALL. Topp MS, Goekbuget N, Stein AS, et al. J Clin Oncol 32:5s, 2014 (suppl; abstr 7005)</s
KEYTRUDA® (Pembrolizumab)
The FDA on September 4, 2014 granted accelerated approval to KEYTRUDA® for the treatment of patients with unresectable or metastatic melanoma and disease progression following YERVOY® (Ipilimumab) and, if BRAF V600 mutation positive, a BRAF inhibitor. KEYTRUDA® is a product of Merck Sharp & Dohme Corp.
KEYTRUDA® – A promising Immunotherapy for Metastatic Melanoma
The FDA granted accelerated approval to KEYTRUDA® (Pembrolizumab), a humanized anti PD-1 antibody, for the treatment of patients with advanced Metastatic Melanoma, who have disease progression following YERVOY® (Ipilimumab) and if BRAF V600 mutation positive, a BRAF inhibitor. KEYTRUDA® produced significant and durable responses in patients with advanced Melanoma, regardless of prior therapy with YERVOY® and this benefit was accomplished with minimal toxicities. This new entry will revolutionize the treatment of advanced Melanoma.
Comparative Outcomes of Catheter-Directed Thrombolysis Plus Anticoagulation vs Anticoagulation Alone to Treat Lower-Extremity Proximal Deep Vein Thrombosis
SUMMARY: The Center for Disease Control and Prevention (CDC) estimates that approximately 1-2 per 1000 individuals develop Deep Vein Thrombosis/Pulmonary Embolism (PE) each year in the United States, resulting in 60,000 – 100,000 deaths. Even though Deep Vein Thrombosis (DVT) commonly occurs in the lower extremities, Non-Leg Deep Venous Thromboses (NLDVT) at other sites including the head and neck, trunk, and upper extremities can occur as well. Approximately one third of the patients with proximal Deep Vein Thrombosis (DVT) develop Post Thrombotic Syndrome which can be associated with pain and swelling in the extremity and if persistent, over time, can lead to skin pigmentation and ulceration, impacting the patients quality of life.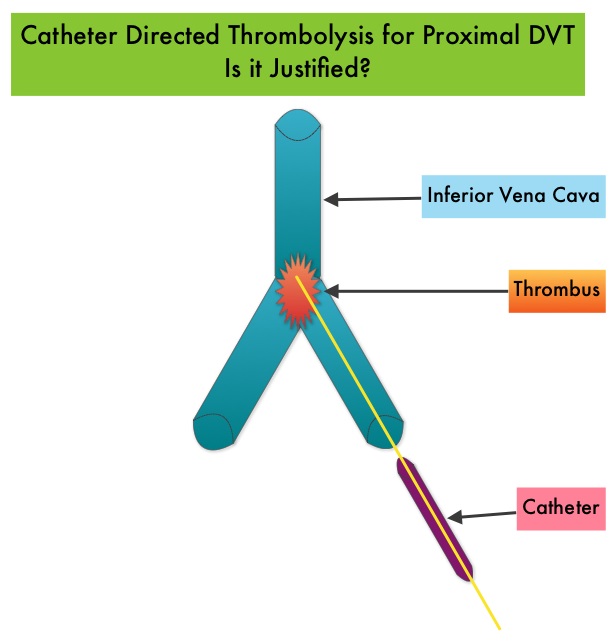 Several small clinical studies have shown a decrease in the incidence of Post Thrombotic Syndrome, with Catheter Directed Thrombolysis (CDT), although safety outcomes from this intervention remained inconclusive. This had resulted in conflicting recommendations from the American Heart Association and American College of Chest Physicians. To address this further, the authors in this study compared safety outcomes in a group of patients with proximal and caval DVT who underwent CDT plus anticoagulation with another group treated with anticoagulation alone. In this propensity-matched analysis, 90,618 patients with a discharge diagnosis of proximal DVT over a 6 year period and 3,594 well-matched patients in each study group were identified from a Nationwide Inpatient Sample (NIS) database. The primary endpoint of this study was in-hospital mortality and secondary endpoints included incidence of pulmonary embolism, blood transfusion requirements, gastrointestinal bleeding, intracranial bleeding, IVC filter placement, procedure-related hematomas, length of hospital stay and hospital charges. This analysis did not demonstrate a significant difference in the in-hospital mortality between patients who had Catheter Directed Thrombolysis (CDT) plus anticoagulation group and the anticoagulation alone group (1.2% vs 0.9%). However, the rates of blood transfusion (11.1% vs 6.5%), pulmonary embolism (17.9% vs 11.4%), intracranial hemorrhage (0.9% vs 0.3%) and IVC filter placement (34.8% vs15.6%), were significantly higher in the CDT group compared to the anticoagulation alone group. Further, patients in the CDT group had a significantly longer hospital stay (7.2 vs. 5.0 days) and incurred significantly higher hospital expenses ($85,094 vs $28,164). More patients with private insurance had Catheter Directed Thrombolysis (CDT), compared to self-pay patients or those with Medicare or Medicaid (7.3% vs 2.8%, P<0.001). The authors concluded that Catheter Directed Thrombolysis (CDT) may not improve outcomes for patients with proximal Deep Vein Thrombosis but is associated with significantly higher adverse events and therefore warrants substantial justification, if this intervention is planned. Bashir R, Zack CJ, Zhao H, et al. JAMA Intern Med. 2014;174:1494-1501
Several small clinical studies have shown a decrease in the incidence of Post Thrombotic Syndrome, with Catheter Directed Thrombolysis (CDT), although safety outcomes from this intervention remained inconclusive. This had resulted in conflicting recommendations from the American Heart Association and American College of Chest Physicians. To address this further, the authors in this study compared safety outcomes in a group of patients with proximal and caval DVT who underwent CDT plus anticoagulation with another group treated with anticoagulation alone. In this propensity-matched analysis, 90,618 patients with a discharge diagnosis of proximal DVT over a 6 year period and 3,594 well-matched patients in each study group were identified from a Nationwide Inpatient Sample (NIS) database. The primary endpoint of this study was in-hospital mortality and secondary endpoints included incidence of pulmonary embolism, blood transfusion requirements, gastrointestinal bleeding, intracranial bleeding, IVC filter placement, procedure-related hematomas, length of hospital stay and hospital charges. This analysis did not demonstrate a significant difference in the in-hospital mortality between patients who had Catheter Directed Thrombolysis (CDT) plus anticoagulation group and the anticoagulation alone group (1.2% vs 0.9%). However, the rates of blood transfusion (11.1% vs 6.5%), pulmonary embolism (17.9% vs 11.4%), intracranial hemorrhage (0.9% vs 0.3%) and IVC filter placement (34.8% vs15.6%), were significantly higher in the CDT group compared to the anticoagulation alone group. Further, patients in the CDT group had a significantly longer hospital stay (7.2 vs. 5.0 days) and incurred significantly higher hospital expenses ($85,094 vs $28,164). More patients with private insurance had Catheter Directed Thrombolysis (CDT), compared to self-pay patients or those with Medicare or Medicaid (7.3% vs 2.8%, P<0.001). The authors concluded that Catheter Directed Thrombolysis (CDT) may not improve outcomes for patients with proximal Deep Vein Thrombosis but is associated with significantly higher adverse events and therefore warrants substantial justification, if this intervention is planned. Bashir R, Zack CJ, Zhao H, et al. JAMA Intern Med. 2014;174:1494-1501
Clinical Impact of Delaying Initiation of Adjuvant Chemotherapy in Patients with Breast Cancer
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately, 233,000 new cases of invasive breast cancer will be diagnosed in 2014 and 40,000 women will die of the disease. The HER or erbB family of receptors consist of HER1, HER2, HER3 and HER4. Approximately 15%-20% of invasive breast cancers overexpress HER2/neu oncogene, which is a negative predictor of outcomes, without systemic therapy. Patients with early stage breast cancer often receive adjuvant chemotherapy and this is even more so true for HER positive and triple negative (ER, PR and HER negative) breast cancer patients, who are at an increased risk to develop recurrent disease. Even though majority of the patients start their adjuvant chemotherapy within 4-6 weeks following surgery, the impact of delay in the initiation of adjuvant therapy, on outcomes, has remained unclear. Preclinical models have suggested that there is phase of increased angiogenesis and accelerated growth of micrometastases, as well as development of drug resistant clones, following removal of the primary tumor. Previously published data from a large meta-analysis had suggested that a four week delay in the initiation of adjuvant chemotherapy resulted in a 6% increase in the risk of death and an 8% increase in the risk of relapse. Based on this background information, the authors in this study evaluated the impact of time to initiation of adjuvant chemotherapy, on survival, in patients with various stages and subtypes of early stage breast cancer. In this single institution study, 6,827 women diagnosed with stages I to III breast cancer between 1997 and 2011, were categorized into one of three groups – 30 days or less, 31 to 60 days and 61 days or more, according to the time from definitive surgery to adjuvant chemotherapy. Survival outcomes were then estimated in these three groups. The median follow up was 59.3 months and majority of the patients (84.5%) had stage I or II breast cancer and 15.5% of the patients had stage III disease. The authors noted that outcomes were inferior among patients with stage II and stage III disease when chemotherapy was initiated 61 days or more after surgery, with a 76% increase in the risk of death among patients with stage III disease. This disadvantage was however not noted in patients with stage I disease. Survival estimates based on the tumor sub types revealed that patients with triple negative breast cancer tumors and those with HER-2 positive (Human Epidermal growth factor Receptor- 2) tumors, treated 61 days or more after surgery with HERCEPTIN® (Trastuzumab) based chemotherapy, had the worse survival, compared with those who initiated adjuvant treatment within the first 30 days after surgery. Patients with hormone receptor positive tumors were however not impacted. The authors concluded that delaying the initiation of adjuvant chemotherapy in patients with high risk disease such as those with Stages II and III breast cancer and those with triple negative breast cancer and HER-2 positive tumors, can negatively impact survival outcomes. de Melo Gagliato D, Gonzalez-Angulo AM, Lei X, et al. J Clin Oncol 2014;32:735-744
Panorama 1 A randomized, double-blind, phase 3 study of panobinostat or placebo plus bortezomib and dexamethasone in relapsed or relapsed and refractory multiple myeloma
SUMMARY:Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, 24,050 new cases will be diagnosed in 2014 and 11,090 will die of the disease. The authors in the PANORAMA I trial evaluated the outcomes in previously treated advanced multiple myeloma patients, by taking advantage of the synergy between Bortezomib (VELCADE®), a proteosome inhibitor and Panobinostat, a histone deacetylase (HDAC) inhibitor and treating these patients with a combination of these two agents. HDACs are a family of enzymes that play an important role in the regulation of gene expression.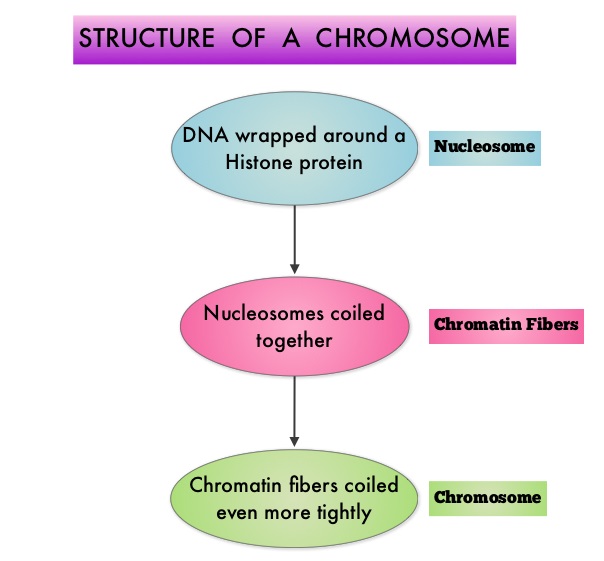 To briefly summarize the structure of a chromosome, individual loops of coiled double-helix DNA wrap around a histone protein to form a nucleosome. Nucleosomes are then coiled together to form chromatin fibers, which looks like beads on a string. The chromatin fibers are coiled even more tightly to form chromosomes. HDAC enzymes catalyze the removal of acetyl groups and regulate the level of acetylation of the histones and non-histone proteins and transcription of several genes. Hypoacetylation of histones has been associated with a condensed chromatin structure that results in the repression of gene transcription, whereas acetylated histones are associated with a more open chromatin structure and activation of gene transcription. HDACs are grouped into four major classes (Class I, II, III and IV) and regulate cell-cycle progression, cell survival, angiogenesis and immunity. The HDAC Class I enzymes are HDAC1, 2, 3 & 8 and are typically found in the nucleus where they are able to repress transcription. The HDAC Class II enzymes include HDAC4, 5, 6, 7, 9 and 10 and are able to move between the cytoplasm and nucleus and function in signal transduction. In Multiple Myeloma, the important enzyme to target is HDAC6. Panobinostat is an oral, pan-histone deacetylase inhibitor which inhibits cell cycle progression and ultimately results in apoptosis. Panobinostat inhibits the aggresome pathway of protein degradation which is upregulated when proteosome pathway is inhibited by VELCADE®.
To briefly summarize the structure of a chromosome, individual loops of coiled double-helix DNA wrap around a histone protein to form a nucleosome. Nucleosomes are then coiled together to form chromatin fibers, which looks like beads on a string. The chromatin fibers are coiled even more tightly to form chromosomes. HDAC enzymes catalyze the removal of acetyl groups and regulate the level of acetylation of the histones and non-histone proteins and transcription of several genes. Hypoacetylation of histones has been associated with a condensed chromatin structure that results in the repression of gene transcription, whereas acetylated histones are associated with a more open chromatin structure and activation of gene transcription. HDACs are grouped into four major classes (Class I, II, III and IV) and regulate cell-cycle progression, cell survival, angiogenesis and immunity. The HDAC Class I enzymes are HDAC1, 2, 3 & 8 and are typically found in the nucleus where they are able to repress transcription. The HDAC Class II enzymes include HDAC4, 5, 6, 7, 9 and 10 and are able to move between the cytoplasm and nucleus and function in signal transduction. In Multiple Myeloma, the important enzyme to target is HDAC6. Panobinostat is an oral, pan-histone deacetylase inhibitor which inhibits cell cycle progression and ultimately results in apoptosis. Panobinostat inhibits the aggresome pathway of protein degradation which is upregulated when proteosome pathway is inhibited by VELCADE®.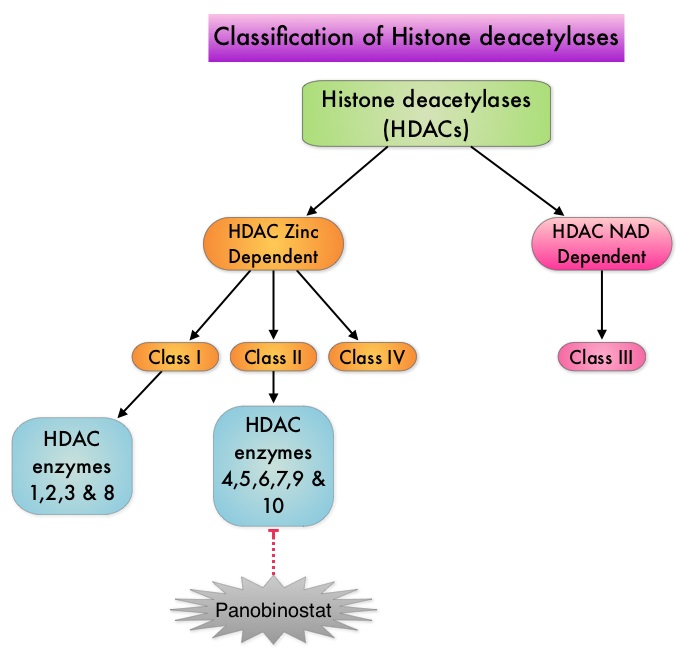 Based on preclinical data demonstrating synergy between VELCADE® and Panobinostat in Myeloma, the PANORAMA 1 trial, enrolled patients with relapsed or refractory multiple myeloma who had received one to three prior lines of therapy and were not VELCADE® refractory. In this phase III trial, patients were randomly assigned to receive either Panobinostat (N=387) or Placebo (N=381), each along with IV VELCADE® and oral Dexamethasone. For cycles 1 thru 8, patients received Panobinostat 20 mg PO or Placebo on days 1, 3, 5, 8, 10, and 12; VELCADE® 1.3 mg/m2 IV on days 1, 4, 8, and 11; and Dexamethasone 20 mg PO on days 1-2, 4-5, 8-9, and 11-12 of a 21 day cycle. Patients with clinical benefit after the first eight cycles could proceed to the second phase of treatment in which VELCADE® was administered only on D1 and D8 and Dexamethasone administered only on days 1-2 and 8-9. The median age was 63 years, 48% of patients had received at least two lines of therapy and 57% of patients had prior autologous stem cell transplantation and 43% had prior therapy with VELCADE®. The primary endpoint was Progression Free Survival (PFS) and secondary endpoints included Overall Survival (OS), Overall Response Rate (ORR), near Complete/Complete Response (nCR/CR) rate, Duration of Response (DOR), and safety. After a median follow up of 28 months, the primary end point of the study was met with a 37% decrease in the risk of disease progression in the Panobinostat group compared to the Placebo group (12 months vs 8.1 months, HR=0.63, P<0.0001). With regards to the secondary endpoints in the Panobinostat vs Placebo groups, the ORR was 60.7% vs 54.6% (P=0.87), nCR/CR rate was 27.6% vs 15.7% (P=0.00006), median duration of response was13.1months vs 10.9 months and median time to progression was 12.7 months vs 8.5 months respectively. The most common grade 3/4 adverse events in the Panobinostat vs Placebo arms included thrombocytopenia (67% vs 31%), neutropenia (35% vs 11%), and diarrhea (26% vs 8%) and these toxicities were manageable with dose reduction and supportive care. The authors concluded that a combination of Panobinostat, VELCADE® and Dexamethasone significantly improves Progression Free Survival in patients with relapsed and refractory Multiple Myeloma, with manageable toxicities. Richardson PG, Hungria VTM , Yoon S, et al. J Clin Oncol 32:5s, 2014 (suppl; abstr 8510)</s
Based on preclinical data demonstrating synergy between VELCADE® and Panobinostat in Myeloma, the PANORAMA 1 trial, enrolled patients with relapsed or refractory multiple myeloma who had received one to three prior lines of therapy and were not VELCADE® refractory. In this phase III trial, patients were randomly assigned to receive either Panobinostat (N=387) or Placebo (N=381), each along with IV VELCADE® and oral Dexamethasone. For cycles 1 thru 8, patients received Panobinostat 20 mg PO or Placebo on days 1, 3, 5, 8, 10, and 12; VELCADE® 1.3 mg/m2 IV on days 1, 4, 8, and 11; and Dexamethasone 20 mg PO on days 1-2, 4-5, 8-9, and 11-12 of a 21 day cycle. Patients with clinical benefit after the first eight cycles could proceed to the second phase of treatment in which VELCADE® was administered only on D1 and D8 and Dexamethasone administered only on days 1-2 and 8-9. The median age was 63 years, 48% of patients had received at least two lines of therapy and 57% of patients had prior autologous stem cell transplantation and 43% had prior therapy with VELCADE®. The primary endpoint was Progression Free Survival (PFS) and secondary endpoints included Overall Survival (OS), Overall Response Rate (ORR), near Complete/Complete Response (nCR/CR) rate, Duration of Response (DOR), and safety. After a median follow up of 28 months, the primary end point of the study was met with a 37% decrease in the risk of disease progression in the Panobinostat group compared to the Placebo group (12 months vs 8.1 months, HR=0.63, P<0.0001). With regards to the secondary endpoints in the Panobinostat vs Placebo groups, the ORR was 60.7% vs 54.6% (P=0.87), nCR/CR rate was 27.6% vs 15.7% (P=0.00006), median duration of response was13.1months vs 10.9 months and median time to progression was 12.7 months vs 8.5 months respectively. The most common grade 3/4 adverse events in the Panobinostat vs Placebo arms included thrombocytopenia (67% vs 31%), neutropenia (35% vs 11%), and diarrhea (26% vs 8%) and these toxicities were manageable with dose reduction and supportive care. The authors concluded that a combination of Panobinostat, VELCADE® and Dexamethasone significantly improves Progression Free Survival in patients with relapsed and refractory Multiple Myeloma, with manageable toxicities. Richardson PG, Hungria VTM , Yoon S, et al. J Clin Oncol 32:5s, 2014 (suppl; abstr 8510)</s
Electronic Cigarettes A Position Statement of the Forum of International Respiratory Societies
SUMMARY: According to the American Cancer Society, tobacco use is responsible for nearly 1 in 5 deaths in the United States and accounts for at least 30% of all cancer deaths. Smokeless tobacco products are a major source of cancer causing nitrosamines and increase the risk of developing cancer of the oropharynx, esophagus, and pancreas. Cigarette smoke contains more than 7,000 chemicals, many of which are toxic and some linked to cancer. E-cigarettes were first developed in China and were introduced to the U.S. market in 2007. When a smoker inhales through the mouth piece of an E-cigarette, the air flow triggers a sensor that switches on a small lithium battery powered heater, which in turn vaporizes liquid nicotine along with PolyEthylene Glycol (PEG) present in a small cartridge. The PEG vapor looks like smoke. The potent liquid form of nicotine extracted from tobacco is tinctured with fragrant flavors such as chocolate, cherry and bubble gum, coloring substances, as well other chemicals and these e-liquids are powerful neurotoxins. With the rapid growth of the E-cigarette industry and the evidence of potential dangers and risk to public health, particularly children, experts from the world's leading lung organizations were compelled to release a position statement on electronic cigarettes, specifically focusing on their potential adverse effects on human health and calling on government organizations to ban or restrict the use of E-cigarettes, until their impact on health is better understood. With epidemiological data demonstrating that nicotine use is a gateway to the use of cocaine and marijuana and subsequent lifelong addiction, the Forum of International Respiratory Societies (FIRS), an organization composed of the world's leading international respiratory societies including American Thoracic Society (ATS) and the American College of Chest Physicians (ACCP), made the following recommendations. The position statement of the Forum of International Respiratory Societies (FIRS) on electronic nicotine delivery devices includes the following:
When a smoker inhales through the mouth piece of an E-cigarette, the air flow triggers a sensor that switches on a small lithium battery powered heater, which in turn vaporizes liquid nicotine along with PolyEthylene Glycol (PEG) present in a small cartridge. The PEG vapor looks like smoke. The potent liquid form of nicotine extracted from tobacco is tinctured with fragrant flavors such as chocolate, cherry and bubble gum, coloring substances, as well other chemicals and these e-liquids are powerful neurotoxins. With the rapid growth of the E-cigarette industry and the evidence of potential dangers and risk to public health, particularly children, experts from the world's leading lung organizations were compelled to release a position statement on electronic cigarettes, specifically focusing on their potential adverse effects on human health and calling on government organizations to ban or restrict the use of E-cigarettes, until their impact on health is better understood. With epidemiological data demonstrating that nicotine use is a gateway to the use of cocaine and marijuana and subsequent lifelong addiction, the Forum of International Respiratory Societies (FIRS), an organization composed of the world's leading international respiratory societies including American Thoracic Society (ATS) and the American College of Chest Physicians (ACCP), made the following recommendations. The position statement of the Forum of International Respiratory Societies (FIRS) on electronic nicotine delivery devices includes the following:
• The safety of electronic cigarettes has not been adequately demonstrated.
• The addictive power of nicotine and its untoward effects should not be underestimated.
• The potential benefits of electronic nicotine delivery devices, including harm reduction and as an aid to smoking cessation, have not been well studied.
• Potential benefits to an individual smoker should be weighed against harm to the population of increased social acceptability of smoking and use of nicotine.
• Health and safety claims regarding electronic nicotine delivery devices should be subject to evidentiary review.
• Adverse health effects for third parties exposed to the emissions of electronic cigarettes cannot be excluded.
• Electronic nicotine delivery devices should be restricted or banned, at least until more information about their safety is available.
• If electronic nicotine delivery devices are permitted, they should be regulated as medicines and subject to the same evidentiary review of other medicines.
• If electronic nicotine delivery devices are not regulated as medicines, they should be regulated as tobacco products.
• Research, supported by sources other than the tobacco or electronic cigarette industry, should be carried out to determine the impact of electronic nicotine delivery devices on health in a wide variety of settings.
• The use and population effects of electronic nicotine delivery devices should be monitored.
• All information derived from this research should be conveyed to the public in a clear manner.
Schraufnagel DE, Blasi F, Drummond MB, et al. on behalf of the Forum of International Respiratory Societies. Am J Respir Crit Care Med. First published online 09 Jul 2014 as DOI: 10.1164/rccm.201407-1198PP