
SUMMARY: It is estimated that in the US, approximately 76,000 new cases of melanoma were diagnosed and close to 8000 individuals died of the disease, in 2014. The incidence of melanoma has been on the rise for the past three decades. The FDA granted accelerated approval in January 2014, for a combination of MEKINIST® (Trametinib) and TAFINLAR® (Dabrafenib), to treat patients with advanced melanoma, based on the understanding of the biological pathways of this malignancy. The Mitogen-Activated Protein Kinase pathway (MAPK pathway) is an important signaling pathway which enables the cell to respond to external stimuli. This pathway plays a dual role regulating cytokine production and participating in cytokine dependent signaling cascade. The MAPK pathway of interest is the RAS-RAF-MEK-ERK pathway. The RAF family of kinases includes ARAF, BRAF and CRAF signaling molecules. BRAF is a very important intermediary of the RAS-RAF-MEK-ERK pathway. BRAF mutations have been demonstrated in 6%-8% of all malignancies. The most common BRAF mutation in melanoma is at the V600E/K site and is detected in approximately 50% of melanomas. In the BREAK-3 randomized phase III trial, TAFINLAR® (Dabrafenib), a selective oral BRAF inhibitor demonstrated a statistically significant improvement in Progression Free Survival (PFS) and Response Rate (RR) compared to Dacarbazine (DTIC), in patients with advanced BRAF V600E/K mutated melanoma. In the BRIM 3 randomized, phase III study, ZELBORAF® (Vemurafenib), a selective oral inhibitor of mutated BRAF demonstrated significant improvement in Progression Free Survival and Overall Survival compared to Dacarbazine.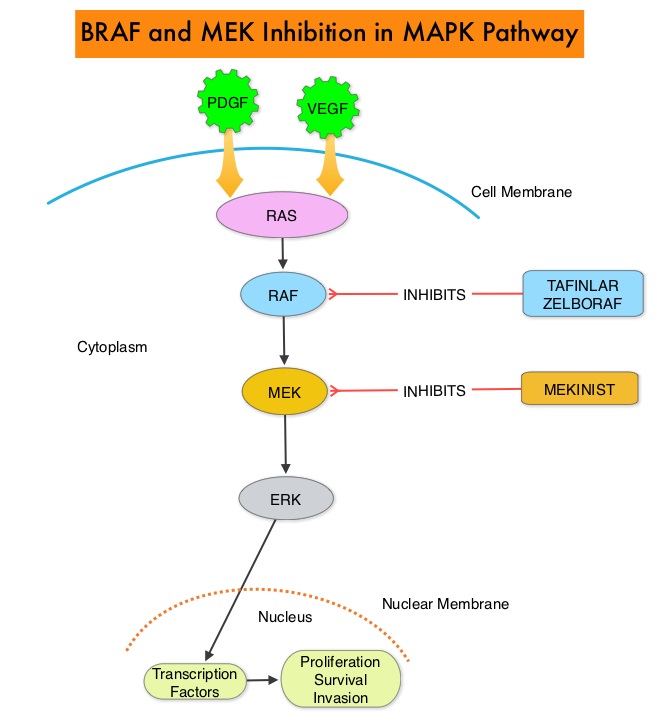 Squamous cell carcinoma’s were seen in about 6% of the patients treated with BRAF inhibitors. Paradoxical activation of the MAPK pathway in cells without a BRAF mutation has been implicated in the emergence of drug resistance and increased incidence of BRAF-inhibitor induced skin tumors. The addition of a MEK inhibitor such as MEKINIST® (Trametinib) to a BRAF inhibitor such as TAFINLAR®, has addressed some of these limitations, in previously published studies, with improvement in Progression Free Survival. MEKINIST® is a potent and selective inhibitor of MEK gene, which is downstream from RAF in the MAPK pathway and has been shown to significantly improve Progression Free Survival, Response Rate and Overall Survival, when compared to chemotherapy, in advanced melanoma patients with BRAF V600E/K mutations. The authors in this open label, randomized, phase III trial, evaluated the outcomes, comparing a combination of TAFINLAR® and MEKINIST® with single agent ZELBORAF® , in previously untreated and unresectable Stage IIIC or IV melanoma patients with BRAF V600E or V600K mutations. Eligible patients (N=704) were assigned in a 1:1 ratio to receive either a combination of TAFINLAR® (Dabrafenib), 150 mg PO BID and MEKINIST ® (Trametinib) 2 mg PO QD or ZELBORAF® (Vemurafenib) 960 mg PO BID, as first line therapy. The primary end point of this study was Overall Survival. Secondary end points included Progression Free Survival, Overall Response Rate, duration of response, and safety. At the preplanned interim analysis, the Overall Survival at 12 months was 72% in the combination therapy group and 65% in the single agent ZELBORAF® group (HR=0.69; P=0.005). The median Progression Free Survival was 11.4 months in the combination therapy group and 7.3 months in the ZELBORAF® group (HR=0.56; P<0.001). The objective response rate was 64% with combination therapy and 51% with single agent ZELBORAF® (P<0.001). There was no difference in the rates of severe adverse events and study drug discontinuations between the two groups. Skin cancers such as Squamous cell carcinoma and Keratoacanthoma occurred in 1% of patients in the combination therapy group and 18% of those treated with ZELBORAF® . The authors concluded that a combination of BRAF inhibitor TAFINLAR® and MEK inhibitor MEKINIST® significantly improved Overall Survival as compared with ZELBORAF® monotherapy, with a 31% relative reduction in the risk of death, in previously untreated patients with metastatic melanoma, with BRAF V600E or V600K mutations. This benefit was accomplished without increased overall toxicity. Robert C, Karaszewska B, Schachter J, et al. N Engl J Med 2015; 372:30-39
Squamous cell carcinoma’s were seen in about 6% of the patients treated with BRAF inhibitors. Paradoxical activation of the MAPK pathway in cells without a BRAF mutation has been implicated in the emergence of drug resistance and increased incidence of BRAF-inhibitor induced skin tumors. The addition of a MEK inhibitor such as MEKINIST® (Trametinib) to a BRAF inhibitor such as TAFINLAR®, has addressed some of these limitations, in previously published studies, with improvement in Progression Free Survival. MEKINIST® is a potent and selective inhibitor of MEK gene, which is downstream from RAF in the MAPK pathway and has been shown to significantly improve Progression Free Survival, Response Rate and Overall Survival, when compared to chemotherapy, in advanced melanoma patients with BRAF V600E/K mutations. The authors in this open label, randomized, phase III trial, evaluated the outcomes, comparing a combination of TAFINLAR® and MEKINIST® with single agent ZELBORAF® , in previously untreated and unresectable Stage IIIC or IV melanoma patients with BRAF V600E or V600K mutations. Eligible patients (N=704) were assigned in a 1:1 ratio to receive either a combination of TAFINLAR® (Dabrafenib), 150 mg PO BID and MEKINIST ® (Trametinib) 2 mg PO QD or ZELBORAF® (Vemurafenib) 960 mg PO BID, as first line therapy. The primary end point of this study was Overall Survival. Secondary end points included Progression Free Survival, Overall Response Rate, duration of response, and safety. At the preplanned interim analysis, the Overall Survival at 12 months was 72% in the combination therapy group and 65% in the single agent ZELBORAF® group (HR=0.69; P=0.005). The median Progression Free Survival was 11.4 months in the combination therapy group and 7.3 months in the ZELBORAF® group (HR=0.56; P<0.001). The objective response rate was 64% with combination therapy and 51% with single agent ZELBORAF® (P<0.001). There was no difference in the rates of severe adverse events and study drug discontinuations between the two groups. Skin cancers such as Squamous cell carcinoma and Keratoacanthoma occurred in 1% of patients in the combination therapy group and 18% of those treated with ZELBORAF® . The authors concluded that a combination of BRAF inhibitor TAFINLAR® and MEK inhibitor MEKINIST® significantly improved Overall Survival as compared with ZELBORAF® monotherapy, with a 31% relative reduction in the risk of death, in previously untreated patients with metastatic melanoma, with BRAF V600E or V600K mutations. This benefit was accomplished without increased overall toxicity. Robert C, Karaszewska B, Schachter J, et al. N Engl J Med 2015; 372:30-39
Month: January 2015
p16 Protein Expression and Human Papillomavirus Status As Prognostic Biomarkers of Non-Oropharyngeal Head and Neck Squamous Cell Carcinoma
SUMMARY: Squamous Cell Carcinoma of the Head and Neck (HNSCC) accounts for about 3-5% of all cancers in the United States. It is estimated that 55,070 people were diagnosed with Head and Neck cancer in 2014 and 12,000 patients died of the disease. The head and neck region includes the oral cavity, oropharynx, hypopharynx and larynx. Squamous Cell Carcinoma involving the head and neck region (HNSCC) is therefore a heterogeneous disease and the five year survival rate of patients with head and neck cancer depends on several factors and can be variable. Common risk factors include tobacco and alcohol use and Human PapillomaVirus (HPV) infection. Recent studies have shown that over 70% of the OroPharyngeal Squamous Cell Carcinomas (OPSCC) are caused by HPV with the HPV-16 being the predominant type present in the tumor cells.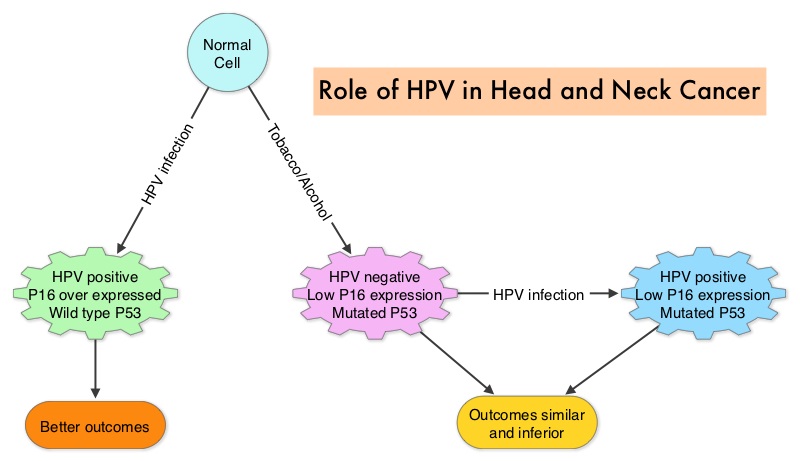 The malignant behavior of these tumors is dependent on the expression of viral E6 and E7 oncoproteins that inactivate the tumor suppressor proteins p53 and the retinoblastoma protein (pRB), respectively. HPV positive OroPharyngeal Squamous Cell Carcinoma (OPSCC) is more common among never smokers or light smokers and patients tend to be younger with better performance status. The pathobiology of HPV positive OroPharyngeal Squamous Cell Carcinoma (OPSCC) is different from the HPV negative OroPharyngeal Squamous Cell Carcinoma in that p53 is degraded/inactivated by viral E6 oncoprotein instead of by genetic mutation, pRB pathway is inactivated by viral E7 oncoprotein instead of Cyclin D1 amplification and p16 is overexpressed or upregulated instead of inactivation resulting from reduced negative feedback from pRB. Testing for HPV is based on several methodologies which include detection of HPV DNA by in situ hybridization (ISH) or Polymerase Chain Reaction (PCR), detection of HPV E6/E7 RNA expression by quantitative Reverse Transcriptase–PCR (qRT-PCR) and p16 protein overexpression, a surrogate marker of oncogenic HPV infection, by ImmunoHistoChemistry (IHC) staining. Both p16 overexpression testing by IHC and HPV by ISH can be easily performed on Formalin-Fixed Paraffin Embedded (FFPE) specimen whereas detection of HPV E6/E7 RNA expression, which is indicative of active viral oncogene transcription in tumor cells and is considered to be a gold standard, requires a larger tumor specimen compared with other methodologies.
The malignant behavior of these tumors is dependent on the expression of viral E6 and E7 oncoproteins that inactivate the tumor suppressor proteins p53 and the retinoblastoma protein (pRB), respectively. HPV positive OroPharyngeal Squamous Cell Carcinoma (OPSCC) is more common among never smokers or light smokers and patients tend to be younger with better performance status. The pathobiology of HPV positive OroPharyngeal Squamous Cell Carcinoma (OPSCC) is different from the HPV negative OroPharyngeal Squamous Cell Carcinoma in that p53 is degraded/inactivated by viral E6 oncoprotein instead of by genetic mutation, pRB pathway is inactivated by viral E7 oncoprotein instead of Cyclin D1 amplification and p16 is overexpressed or upregulated instead of inactivation resulting from reduced negative feedback from pRB. Testing for HPV is based on several methodologies which include detection of HPV DNA by in situ hybridization (ISH) or Polymerase Chain Reaction (PCR), detection of HPV E6/E7 RNA expression by quantitative Reverse Transcriptase–PCR (qRT-PCR) and p16 protein overexpression, a surrogate marker of oncogenic HPV infection, by ImmunoHistoChemistry (IHC) staining. Both p16 overexpression testing by IHC and HPV by ISH can be easily performed on Formalin-Fixed Paraffin Embedded (FFPE) specimen whereas detection of HPV E6/E7 RNA expression, which is indicative of active viral oncogene transcription in tumor cells and is considered to be a gold standard, requires a larger tumor specimen compared with other methodologies.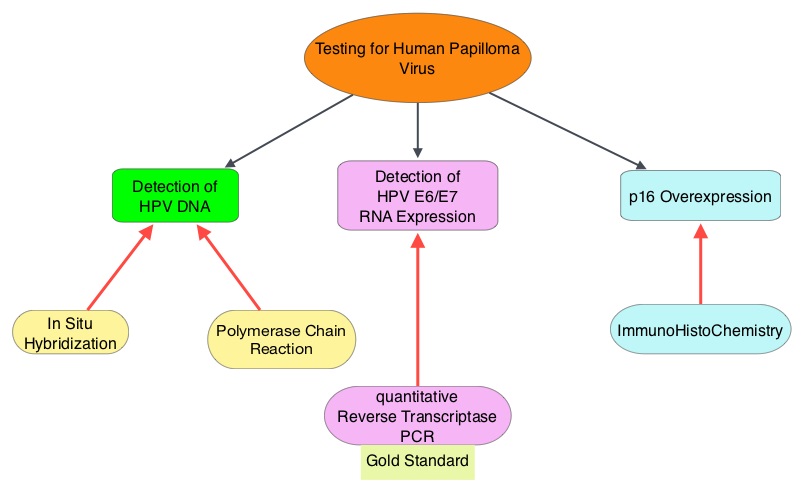 The concordance rate between HPV by ISH and p16 by IHC is approximately 90% in OPSCC, whereas the prevalence and significance p16 by IHC has remained unclear for HPV infection in non-OPSCC (oral cavity, hypopharynx and larynx). It is well established however that patients with HPV-positive/p16-positive OPSCC have better outcomes compared with those with HPV-negative/p16-negative OPSCC. However, the prognostic significance of p16 overexpression in non-OPSCC patients with or without evidence of HPV infection has not been clear. The authors in this study evaluated p16 protein overexpression by IHC and HPV status by HPV ISH as potential prognostic biomarkers in non-OPSCC tumors of patients enrolled in three prospective Radiation Therapy Oncology Group (RTOG) clinical trials. Of the 1921 patient enrolled in these three trials, 683 patients with non-OPSCC tumors were eligible and 52% (N=356) of these patients were tested for p16 overexpression and overall, 19.3% were p16 positive. When OPSCC and non-OPSCC patients were compared, OPSCC patients whose tumors were p16 positive had better Progression Free Survival and Overall Survival than non-OPSCC patients with p16 positive tumors. However, patients with p16 negative OPSCC and non-OPSCC have similar inferior outcomes. The authors concluded that patients with p16 negative non-OPSCC have worse outcomes than patients with p16 positive non-OPSCC, similar to that seen in patients with OPSCC and therefore HPV infection may influence outcomes in a subset of patients with non-OPSCC as well. Better methodologies for HPV detection and correlation with p16 expression will help identify “true” HPV infection related non-OPSCC and thereby enable tailored and less intense treatment, for this favorable group of patients. Chung CH, Zhang Q, Kong CS, et al. J Clin Oncol 2014; 32:3930-3938
The concordance rate between HPV by ISH and p16 by IHC is approximately 90% in OPSCC, whereas the prevalence and significance p16 by IHC has remained unclear for HPV infection in non-OPSCC (oral cavity, hypopharynx and larynx). It is well established however that patients with HPV-positive/p16-positive OPSCC have better outcomes compared with those with HPV-negative/p16-negative OPSCC. However, the prognostic significance of p16 overexpression in non-OPSCC patients with or without evidence of HPV infection has not been clear. The authors in this study evaluated p16 protein overexpression by IHC and HPV status by HPV ISH as potential prognostic biomarkers in non-OPSCC tumors of patients enrolled in three prospective Radiation Therapy Oncology Group (RTOG) clinical trials. Of the 1921 patient enrolled in these three trials, 683 patients with non-OPSCC tumors were eligible and 52% (N=356) of these patients were tested for p16 overexpression and overall, 19.3% were p16 positive. When OPSCC and non-OPSCC patients were compared, OPSCC patients whose tumors were p16 positive had better Progression Free Survival and Overall Survival than non-OPSCC patients with p16 positive tumors. However, patients with p16 negative OPSCC and non-OPSCC have similar inferior outcomes. The authors concluded that patients with p16 negative non-OPSCC have worse outcomes than patients with p16 positive non-OPSCC, similar to that seen in patients with OPSCC and therefore HPV infection may influence outcomes in a subset of patients with non-OPSCC as well. Better methodologies for HPV detection and correlation with p16 expression will help identify “true” HPV infection related non-OPSCC and thereby enable tailored and less intense treatment, for this favorable group of patients. Chung CH, Zhang Q, Kong CS, et al. J Clin Oncol 2014; 32:3930-3938
Aspirin for the Prevention of Recurrent Venous Thromboembolism The INSPIRE Collaboration
SUMMARY: The Center for Disease Control and Prevention (CDC) estimates that approximately 1-2 per 1000 individuals develop Deep Vein Thrombosis/Pulmonary Embolism (PE) each year in the United States, resulting in 60,000 – 100,000 deaths. VTE is the third leading cause of cardiovascular mortality with a mortality rate of up to 25% in those with untreated acute pulmonary embolism. Even though the risk of recurrent VTE is high after anticoagulant treatment is discontinued in patients with a first unprovoked venous thromboembolism (10-30% risk of a recurrence within 5 years following cessation of anticoagulation), a significant number of these patients receive therapy for 6-12 months and are not routinely treated with long term anticoagulant therapy. This may be due to bleeding risks, cost of therapy and need for diligent monitoring while on long term anticoagulation. 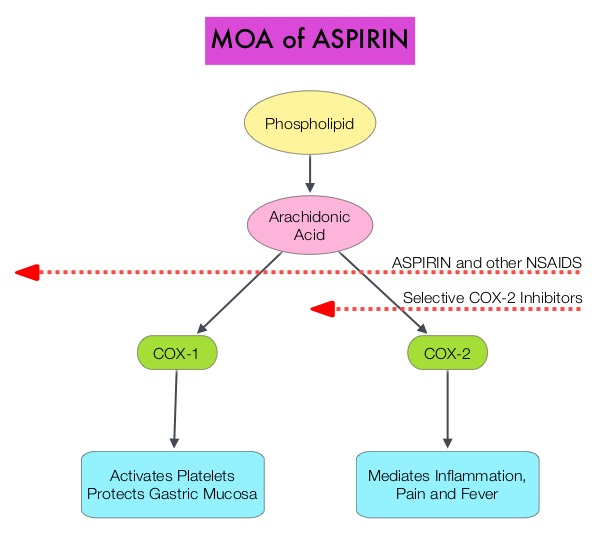 Aspirin is a non-selective, irreversible inhibitor of both COX-1 and COX-2 (Cyclooxygenases 1 and 2). It inhibits the production of Prostaglandins and Thromboxane A2 and there by decreases platelet aggregation. Both ASPIRE and WARFASA trials support the role of aspirin for the prevention of recurrent VTE in patients with an unprovoked VTE, after 6-18 months of anticoagulation therapy. These trials however were not individually powered to detect benefits of treatment for particular outcomes or subgroups of patients. The INSPIRE Collaboration involved further analysis of these trials to detect outcomes in pre-specified subgroups of patients. The intent-to-treat analysis included 1,224 patients and at a median follow up of 30.4 months, daily Aspirin given at a dose of 100mg reduced recurrent VTE events by 32% (HR=0.68; P=0.008) and this included reduction in recurrent Deep Vein Thrombosis by 34% (HR=0.66; P=0.01) and Pulmonary Embolism by 34% (HR=0.66; P=0.08), compared to Placebo. After adjustment for treatment adherence, recurrent VTE was reduced by 42% (HR=0.58; P=0.005). Subgroup analyses indicated similar relative, but larger absolute, risk reductions in men and older patients. Aspirin also reduced major vascular events by 44% (HR=0.66; P=0.002). The major bleeding rate was low at 0.5%/year for Aspirin and 0.4%/year for Placebo. The authors concluded that aspirin reduces the risk of recurrent VTE. Even though the efficacy of aspirin is inferior to that of Warfarin or the new oral anticoagulants, this data suggests that Aspirin should be strongly considered in patients with unprovoked VTE for whom long-term anticoagulation therapy with Warfarin or one of the new oral anticoagulants has to be discontinued or is not an appropriate option. Aspirin however, should not be considered a replacement option for conventional anticoagulation. Simes J, Becattini C, Agnelli G, et al. Circulation. 2014; 130:1062-1071
Aspirin is a non-selective, irreversible inhibitor of both COX-1 and COX-2 (Cyclooxygenases 1 and 2). It inhibits the production of Prostaglandins and Thromboxane A2 and there by decreases platelet aggregation. Both ASPIRE and WARFASA trials support the role of aspirin for the prevention of recurrent VTE in patients with an unprovoked VTE, after 6-18 months of anticoagulation therapy. These trials however were not individually powered to detect benefits of treatment for particular outcomes or subgroups of patients. The INSPIRE Collaboration involved further analysis of these trials to detect outcomes in pre-specified subgroups of patients. The intent-to-treat analysis included 1,224 patients and at a median follow up of 30.4 months, daily Aspirin given at a dose of 100mg reduced recurrent VTE events by 32% (HR=0.68; P=0.008) and this included reduction in recurrent Deep Vein Thrombosis by 34% (HR=0.66; P=0.01) and Pulmonary Embolism by 34% (HR=0.66; P=0.08), compared to Placebo. After adjustment for treatment adherence, recurrent VTE was reduced by 42% (HR=0.58; P=0.005). Subgroup analyses indicated similar relative, but larger absolute, risk reductions in men and older patients. Aspirin also reduced major vascular events by 44% (HR=0.66; P=0.002). The major bleeding rate was low at 0.5%/year for Aspirin and 0.4%/year for Placebo. The authors concluded that aspirin reduces the risk of recurrent VTE. Even though the efficacy of aspirin is inferior to that of Warfarin or the new oral anticoagulants, this data suggests that Aspirin should be strongly considered in patients with unprovoked VTE for whom long-term anticoagulation therapy with Warfarin or one of the new oral anticoagulants has to be discontinued or is not an appropriate option. Aspirin however, should not be considered a replacement option for conventional anticoagulation. Simes J, Becattini C, Agnelli G, et al. Circulation. 2014; 130:1062-1071
Carfilzomib, Lenalidomide, and Dexamethasone for Relapsed Multiple Myeloma
SUMMARY: Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, 24,050 new cases were diagnosed in 2014 and 11,090 died of the disease. KYPROLIS® (Carfilzomib) is a second generation selective, epoxyketone proteasome inhibitor and unlike VELCADE® (Bortezomib), proteasome inhibition with KYPROLIS® is irreversible. KYPROLIS® monotherapy is presently approved in the United States for use in patients with relapsed and refractory Multiple Myeloma following a phase 2b single arm study which showed a 24% overall response rate in this patient group. REVLIMID® (Lenalidomide) given along with weekly Dexamethasone, was associated with significantly improved Progression Free Survival (PFS) when administered until disease progression, in patients with newly diagnosed Multiple Myeloma. The combination of REVLIMID® and weekly Dexamethasone is considered a reference regimen for both newly diagnosed and relapsed multiple myeloma. VELCADE® in combination with REVLIMID® and Dexamethasone showed an overall response rate of 64% and a median PFS of 9.5 months in patients with relapsed and refractory Multiple Myeloma. Based on this background the authors conducted this randomized, open label, multicenter, phase III study in which the safety and efficacy of a combination of KYPROLIS® (Carfilzomib), REVLIMID® and weekly Dexamethasone (KYPROLIS® group) was compared with a combination of REVLIMID® and weekly Dexamethasone (control group), in patients with relapsed Multiple Myeloma. Seven hundred and ninety two (N=792) patients were randomly assigned in a 1:1 ratio to KYPROLIS® group (N=396) and control group (N=396). Eligible patients included those with Multiple Myeloma who had received one to three prior treatments which included VELCADE® or REVLIMID and Dexamethasone combination, provided that they did not have disease progression during treatment with these agents. The 28 day treatment cycle consisted of KYPROLIS® IV given on days 1, 2, 8, 9, 15, and 16 (starting dose, 20 mg/m2 on days 1 and 2 of cycle 1 with a target dose of 27 mg/m2 thereafter) during cycles 1 through 12 and on days 1, 2, 15, and 16 during cycles 13 through 18, following which KYPROLIS® was discontinued. REVLIMID® 25 mg PO was given on days 1 through 21 and Dexamethasone 40 mg PO was administered on days 1, 8, 15, and 22. Patients in both treatment groups received only REVLIMID® and Dexamethasone after cycle 18 until disease progression. Antiviral and antithrombotic prophylaxis was administered to patients in both treatment groups. The primary end point was Progression Free Survival and secondary end points included Overall Survival, the rate of overall response (partial response or better), response duration, health-related quality of life, and safety. The rate of clinical benefit (minimal response or better) was an exploratory end point. The study met its primary endpoint at the time of the pre-specified interim analysis with a significant improvement in the median Progression Free Survival for those patients in the KYPROLIS® group compared to the control group (26.3 months versus 17.6 months; HR=0.69; P=0.0001). This benefit in the PFS was demonstrated across all predefined subgroups. The median overall survival was not reached in either group and the 24 month overall survival rates were 73.3% and 65.0% in the KYPROLIS® and control groups, respectively (HR=0.79; P=0.04). The overall response rates (partial response or better) were 87.1% and 66.7% in the KYPROLIS® and control groups, respectively (P<0.001). Amongst the responders, 31.8% and 9.3% of patients in the respective groups had a complete response or better and 14.1% and 4.3% had a stringent complete response. Further, patients in the KYPROLIS® group reported superior health-related quality of life. Grade 3 or higher adverse events were reported in 83.7% and 80.7% of patients in the KYPROLIS® and control groups respectively. The authors concluded that the addition of KYPROLIS® to REVLIMID® and Dexamethasone resulted in significant improvement in PFS as compared with REVLIMID® and Dexamethasone alone, in patients with relapsed Multiple Myeloma. Additional benefits in the KYPROLIS® group included higher and deep response rates, improved health-related quality of life, a favorable risk–benefit profile and a trend towards improved Overall Survival. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. N Engl J Med 2015; 372:142-152
Adjuvant Paclitaxel and Trastuzumab for Node-Negative, HER2-Positive Breast Cancer
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their life time. Approximately, 233,000 new cases of invasive breast cancer were diagnosed in 2014 and 40,000 women died of the disease. The HER or erbB family of receptors consist of HER1, HER2, HER3 and HER4. Approximately 15%-20% of invasive breast cancers overexpress HER2/neu oncogene, which is a negative predictor of outcomes without systemic therapy. HERCEPTIN® (Trastuzumab) is a humanized monoclonal antibody targeting HER2.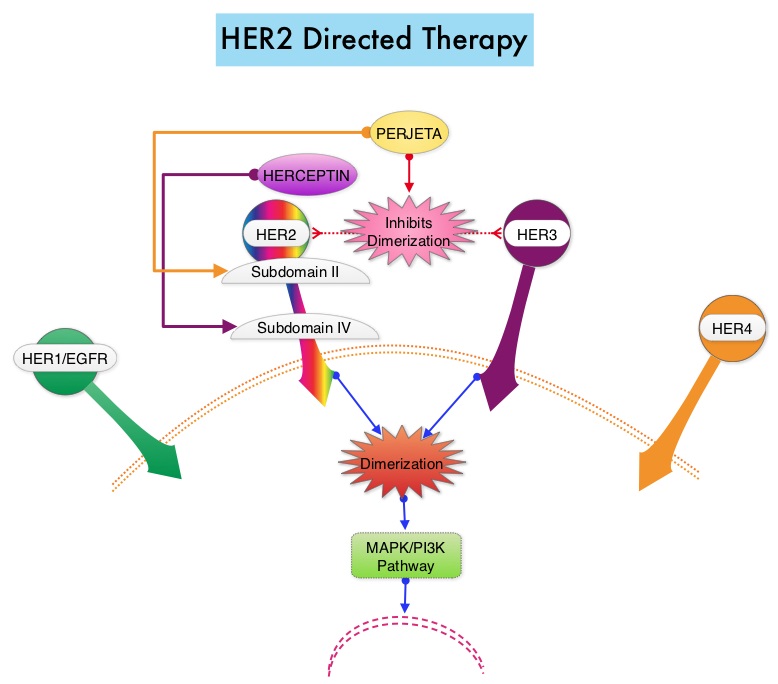 HERCEPTIN® binds to subdomain IV of the HER2 extracellular domain and blocks the downstream cell signaling pathways (PI3K-AKT pathway) and induces Antibody Dependent Cellular Cytotoxicity (ADCC). HERCEPTIN® in combination with chemotherapy has been proven to significantly improve Progression Free Survival and Overall Survival in patients with advanced breast cancer. Adjuvant chemotherapy in combination with HERCEPTIN® has been shown to reduce the relative risk of relapse by 52% and relative risk of death by 33%. The National Comprehensive Cancer Network (NCCN) has recommended adjuvant chemotherapy with HERCEPTIN® for patients with small, HER positive, node-negative tumors, including those with T1bN0 tumors, even though there are little or no data supporting this recommendation, because these patients are generally not included in adjuvant therapy studies. Further, the chemotherapy regimens often recommended (ACTH, TCH) along with HERCEPTIN® are relatively toxic. The authors in this study chose a less toxic chemotherapy regimen than the regimens often recommended for those patients with high risk disease. In this multicenter, investigator initiated study, 406 patients with tumors measuring up to 3 cm in greatest dimension received weekly treatment with TAXOL® (Paclitaxel) and HERCEPTIN (Trastuzumab) for 12 weeks, followed by 9 months of HERCEPTIN® monotherapy. Close to 50% of the patients had tumors 1 cm in diameter or less, about 40% of the patients had tumors 1-2 cm in diameter and majority of the tumors (56%) were high grade. Treatment regimen consisted of TAXOL® 80 mg/m2 IV weekly, for 12 weeks and HERCEPTIN® 4 mg/kg loading dose IV on day 1, followed by 2 mg/kg weekly, for a total of 12 doses followed by HERCEPTIN® 6 mg/kg every 3 weeks for an additional 40 weeks, for a total of 52 weeks of treatment with HERCEPTIN®. Patients who underwent lumpectomy received either partial breast radiation before the initiation of the therapy, or radiation of the whole breast, following completion of treatment with TAXOL®. Treatment with HERCEPTIN® was continued during the time patient was receiving radiation therapy. Adjuvant hormonal therapy was recommended for women with hormone-receptor positive tumors after the completion of TAXOL® treatment. The primary end point was survival free from invasive disease. The median follow up period was 4 years. The 3-year rate of survival free from invasive disease was 98.7%. Treatment was very well tolerated with a low incidence of heart failure (0.5%) and neuropathy. The authors concluded that a less toxic regimen such as HERCEPTIN® given along with weekly TAXOL® has significant efficacy, decreasing the risk of recurrence in this patient group, most notable during the first three years after diagnosis. They also point out that the risk of recurrence of breast cancer is greatest during the first 3-5 years after diagnosis and it would seem unlikely that a different chemotherapy regimen administered with HERCEPTIN® would impact the risk of late recurrences. Tolaney SM, Barry WT, Dang CT, et al. N Engl J Med 2015;372:134-141
HERCEPTIN® binds to subdomain IV of the HER2 extracellular domain and blocks the downstream cell signaling pathways (PI3K-AKT pathway) and induces Antibody Dependent Cellular Cytotoxicity (ADCC). HERCEPTIN® in combination with chemotherapy has been proven to significantly improve Progression Free Survival and Overall Survival in patients with advanced breast cancer. Adjuvant chemotherapy in combination with HERCEPTIN® has been shown to reduce the relative risk of relapse by 52% and relative risk of death by 33%. The National Comprehensive Cancer Network (NCCN) has recommended adjuvant chemotherapy with HERCEPTIN® for patients with small, HER positive, node-negative tumors, including those with T1bN0 tumors, even though there are little or no data supporting this recommendation, because these patients are generally not included in adjuvant therapy studies. Further, the chemotherapy regimens often recommended (ACTH, TCH) along with HERCEPTIN® are relatively toxic. The authors in this study chose a less toxic chemotherapy regimen than the regimens often recommended for those patients with high risk disease. In this multicenter, investigator initiated study, 406 patients with tumors measuring up to 3 cm in greatest dimension received weekly treatment with TAXOL® (Paclitaxel) and HERCEPTIN (Trastuzumab) for 12 weeks, followed by 9 months of HERCEPTIN® monotherapy. Close to 50% of the patients had tumors 1 cm in diameter or less, about 40% of the patients had tumors 1-2 cm in diameter and majority of the tumors (56%) were high grade. Treatment regimen consisted of TAXOL® 80 mg/m2 IV weekly, for 12 weeks and HERCEPTIN® 4 mg/kg loading dose IV on day 1, followed by 2 mg/kg weekly, for a total of 12 doses followed by HERCEPTIN® 6 mg/kg every 3 weeks for an additional 40 weeks, for a total of 52 weeks of treatment with HERCEPTIN®. Patients who underwent lumpectomy received either partial breast radiation before the initiation of the therapy, or radiation of the whole breast, following completion of treatment with TAXOL®. Treatment with HERCEPTIN® was continued during the time patient was receiving radiation therapy. Adjuvant hormonal therapy was recommended for women with hormone-receptor positive tumors after the completion of TAXOL® treatment. The primary end point was survival free from invasive disease. The median follow up period was 4 years. The 3-year rate of survival free from invasive disease was 98.7%. Treatment was very well tolerated with a low incidence of heart failure (0.5%) and neuropathy. The authors concluded that a less toxic regimen such as HERCEPTIN® given along with weekly TAXOL® has significant efficacy, decreasing the risk of recurrence in this patient group, most notable during the first three years after diagnosis. They also point out that the risk of recurrence of breast cancer is greatest during the first 3-5 years after diagnosis and it would seem unlikely that a different chemotherapy regimen administered with HERCEPTIN® would impact the risk of late recurrences. Tolaney SM, Barry WT, Dang CT, et al. N Engl J Med 2015;372:134-141
Hereditary Hemochromatosis Missed Diagnosis or Misdiagnosis?
SUMMARY: It is estimated that 1-6 individuals per 100 in the United States have Iron overload syndrome as measured by random elevated Iron Saturation level. Hereditary Hemochromatosis (HH) is an inherited, Autosomal Recessive, iron storage disease and in the US usually occurs as a result of HFE gene mutations. This is more prevalent among persons of European origin and the HFE gene is located on the short arm of chromosome 6 and modulates iron uptake. Two mutations on the HFE gene, C282Y and H63D, account for the majority of the cases of Hereditary Hemochromatosis, in the United States.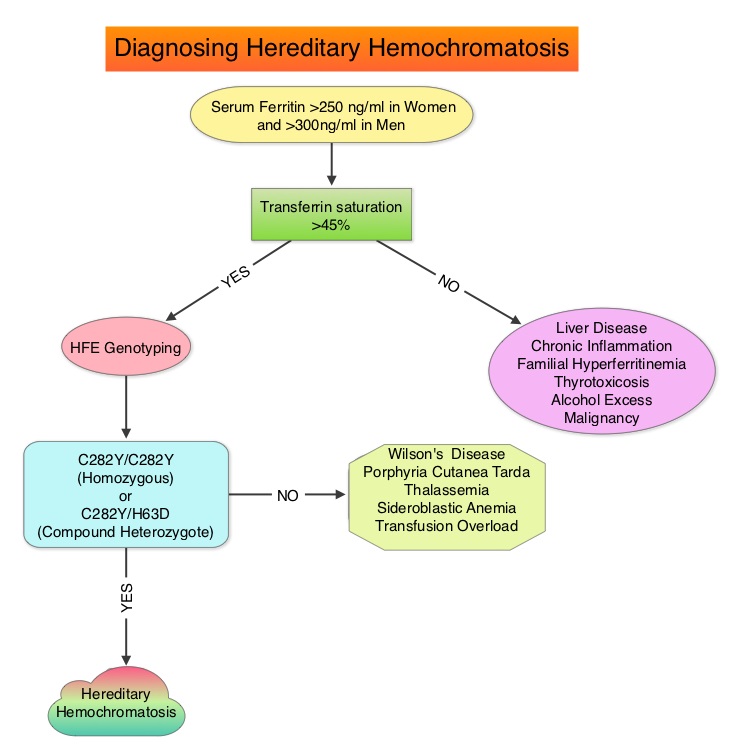 The authors in this provocative study reviewed the electronic medical records of patients seen at a tertiary referral center, to evaluate the accuracy of diagnosis of Hereditary Hemochromatosis (HH). HFE genotyping helps differentiate HH from secondary causes of Iron overload syndromes. This differentiation is relevant because of the lack of established guidelines with regards to management of individuals with abnormal iron studies, secondary to liver disease. It is however well known that HH and iron overload related to transfusions, can cause organ damage and this could be prevented by removing the excess iron. In this study, the authors investigated the diagnostic approach, when elevated iron studies were noted, interpretation of HFE genotyping results by physicians in order to accurately diagnose HH and factors contributing to misdiagnosis. Their review conducted between January 2002 and May 2012, demonstrated that of the 601 patients with disorders of iron metabolism, only 62% were genotyped for mutations in the HFE gene. Of those genotyped, 54% had genotypes consistent with HH (Homozygotes – C282Y/C282Y or Compound Heterozygotes – C282Y/H63D) and the rest of the 46% had non-hereditary hemochromatosis genotypes (C282Y heterozygotes, H63D homozygotes and heterozygotes, etc). One half of these non-hereditary hemochromatosis genotype patients were misdiagnosed as Hereditary Hemochromatosis and a third of these patients underwent phlebotomy. Of those who were not genotyped for mutations in the HFE gene, a third of these patients were diagnosed to have Hereditary Hemochromatosis and majority of these patients underwent phlebotomy. More than two third of the patients misdiagnosed to have Hereditary Hemochromatosis in fact had liver disease and 5% had other hematological conditions. The authors concluded that C282Y heterozygotes as well as H63D homozygotes and heterozygotes do not have Hereditary Hemochromatosis and a majority of patients with iron overload are misdiagnosed to have Hereditary Hemochromatosis. Aggressive phlebotomy in the absence of an appropriate Hereditary Hemochromatosis genotype, is not indicated and can be potentially harmful, delaying more appropriate therapy. Cherfane CE, Hollenbeck RD, Go J, et al. The American Journal of Medicine 2013;126:1010-1015.
The authors in this provocative study reviewed the electronic medical records of patients seen at a tertiary referral center, to evaluate the accuracy of diagnosis of Hereditary Hemochromatosis (HH). HFE genotyping helps differentiate HH from secondary causes of Iron overload syndromes. This differentiation is relevant because of the lack of established guidelines with regards to management of individuals with abnormal iron studies, secondary to liver disease. It is however well known that HH and iron overload related to transfusions, can cause organ damage and this could be prevented by removing the excess iron. In this study, the authors investigated the diagnostic approach, when elevated iron studies were noted, interpretation of HFE genotyping results by physicians in order to accurately diagnose HH and factors contributing to misdiagnosis. Their review conducted between January 2002 and May 2012, demonstrated that of the 601 patients with disorders of iron metabolism, only 62% were genotyped for mutations in the HFE gene. Of those genotyped, 54% had genotypes consistent with HH (Homozygotes – C282Y/C282Y or Compound Heterozygotes – C282Y/H63D) and the rest of the 46% had non-hereditary hemochromatosis genotypes (C282Y heterozygotes, H63D homozygotes and heterozygotes, etc). One half of these non-hereditary hemochromatosis genotype patients were misdiagnosed as Hereditary Hemochromatosis and a third of these patients underwent phlebotomy. Of those who were not genotyped for mutations in the HFE gene, a third of these patients were diagnosed to have Hereditary Hemochromatosis and majority of these patients underwent phlebotomy. More than two third of the patients misdiagnosed to have Hereditary Hemochromatosis in fact had liver disease and 5% had other hematological conditions. The authors concluded that C282Y heterozygotes as well as H63D homozygotes and heterozygotes do not have Hereditary Hemochromatosis and a majority of patients with iron overload are misdiagnosed to have Hereditary Hemochromatosis. Aggressive phlebotomy in the absence of an appropriate Hereditary Hemochromatosis genotype, is not indicated and can be potentially harmful, delaying more appropriate therapy. Cherfane CE, Hollenbeck RD, Go J, et al. The American Journal of Medicine 2013;126:1010-1015.
Adjuvant and Salvage Radiotherapy after Prostatectomy American Society of Clinical Oncology Clinical Practice Guideline Endorsement
SUMMARY: The American Society of Clinical Oncology (ASCO) recently endorsed the Clinical Practice Guidelines recommended by the American Urological Association (AUA)/American Society for Radiation Oncology (ASTRO), on Adjuvant and Salvage Radiotherapy after Prostatectomy. These guidelines target Medical and Radiation Oncologists, Primary care providers, Urologists, other health care providers and address patient counseling, use of radiotherapy in the adjuvant and salvage settings, definition of biochemical recurrence and restaging evaluation. The following are the ASCO Key Recommendations for Adjuvant and Salvage Radiotherapy after Prostatectomy:
1. Patients who are being considered for management of localized prostate cancer with radical prostatectomy should be informed of the potential for adverse pathologic findings that portend a higher risk of cancer recurrence and that these findings may suggest a potential benefit of additional therapy after surgery.
2. Patients with adverse pathologic findings, including seminal vesicle invasion, positive surgical margins, and extraprostatic extension, should be informed that adjuvant radiotherapy, compared with radical prostatectomy only, reduces the risk of biochemical Prostate Specific Antigen (PSA) recurrence, local recurrence, and clinical progression of cancer. They should also be informed that the impact of adjuvant radiotherapy on subsequent metastases and overall survival is less clear; one of two randomized controlled trials that addressed these outcomes indicated a benefit, but the other trial did not demonstrate a benefit defined as reduced risk of metastasis and death.
3. Physicians should “OFFER” adjuvant radiotherapy to patients with adverse pathologic findings at prostatectomy, including seminal vesicle invasion, positive surgical margins, or extraprostatic extension, because of demonstrated reductions in biochemical recurrence, local recurrence and clinical progression.
4. Patients should be informed that the development of a PSA recurrence after surgery is associated with a higher risk of development of metastatic prostate cancer or death resulting from the disease. Congruent with this clinical principle, physicians should regularly monitor PSA after radical prostatectomy to enable early administration of salvage therapies if appropriate.
5. Clinicians should define biochemical recurrence as a detectable or increasing PSA value after surgery that is more than 0.2 ng/mL, with a second confirmatory level more than 0.2 ng/mL.
6. A restaging evaluation in a patient with a PSA recurrence may be considered although it is not clear at this time which imaging modalities to use, as all imaging modalities have limited sensitivity and specificity in the low PSA range.
7. Physicians should “OFFER” salvage radiotherapy to patients with PSA or local recurrence after radical prostatectomy, in whom there is no evidence of distant metastatic disease.
8. Patients should be informed that the effectiveness of radiotherapy for PSA recurrence is greatest when administered at lower levels of PSA (less than 1 ng/ml). Salvage radiotherapy in this patient population with a short PSA doubling time, has been shown to improve overall survival.
9. Patients should be informed of the possible short and long term urinary, bowel, and sexual adverse effects of radiotherapy as well as of the potential benefits of controlling disease recurrence.
This endorsement was made with certain qualifying statements, clarifying certain aspects of these guidelines.
a) The word “OFFER” should be interpreted as having a detailed discussion with the patient about the risks and benefits of adjuvant radiation.
b) Even though 0.2 ng/mL is considered a reasonable cut point for PSA recurrence, the benefits of using this cut point versus other cut points remains unclear.
c) Patient’s who have the greatest benefit in absolute risk reduction from adjuvant RT, are those with adverse pathologic findings as noted in the guidelines, with a high risk of recurrence or clinical progression.
In conclusion, the decision to administer adjuvant or salvage radiotherapy should be made by the patient and multidisciplinary treatment team, after discussing the risks and benefits of such intervention. Freedland SJ, Rumble RB, Finelli A, et al. J Clin Oncol 2014;32:3892-3898
OPDIVO® (Nivolumab)
The FDA on December 22, 2014 granted accelerated approval to OPDIVO® for the treatment of patients with unresectable or metastatic melanoma and disease progression following YERVOY® (Ipilimumab) and if BRAF V600 mutation positive, a BRAF inhibitor. OPDIVO® is a product of Bristol-Myers Squibb Company.
LYNPARZA® (Olaparib)
The FDA on December 19, 2014 approved LYNPARZA® capsules as monotherapy for the treatment of patients with deleterious or suspected deleterious germline BRCA mutated (gBRCAm) (as detected by an FDA-approved test) advanced ovarian cancer, who have been treated with three or more prior lines of chemotherapy. Concurrent with this action, FDA approved the BRACAnalysis CDx® (Myriad Genetics) for the qualitative detection and classification of variants in the BRCA1 and BRCA2 genes. LYNPARZA® capsules is a product of AstraZeneca Pharmaceuticals LP.
SOMATULINE® Depot Injection (Lanreotide)
The FDA on December 16, 2014 approved SOMATULINE® Depot Injection for the treatment of patients with unresectable, well or moderately differentiated, locally advanced or metastatic gastroenteropancreatic neuroendocrine tumors (GEP-NETs) to improve progression-free survival. SOMATULINE® Depot Injection was previously approved for the long-term treatment of acromegalic patients who have had an inadequate response to surgery and/or radiotherapy, or for whom surgery and/or radiotherapy is not an option. SOMATULINE® Depot Injection is a product of Ipsen Pharma.