
SUMMARY: Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, close to 27,000 new cases will be diagnosed in 2015 and 11,240 will die of the disease. The U.S. Food and Drug Administration (FDA) granted accelerated approval on February 23, 2015 to Panobinostat (FARYDAK®), in combination with VELCADE® (Bortezomib) and Dexamethasone, for the treatment of patients with Multiple Myeloma. The authors in the PANORAMA I trial evaluated the outcomes in previously treated advanced Multiple Myeloma patients, by taking advantage of the synergy between VELCADE®, a proteosome inhibitor and FARYDAK® (Panobinostat), a histone deacetylase (HDAC) inhibitor and treating these patients with a combination of these two agents. HDACs are a family of enzymes that play an important role in the regulation of gene expression.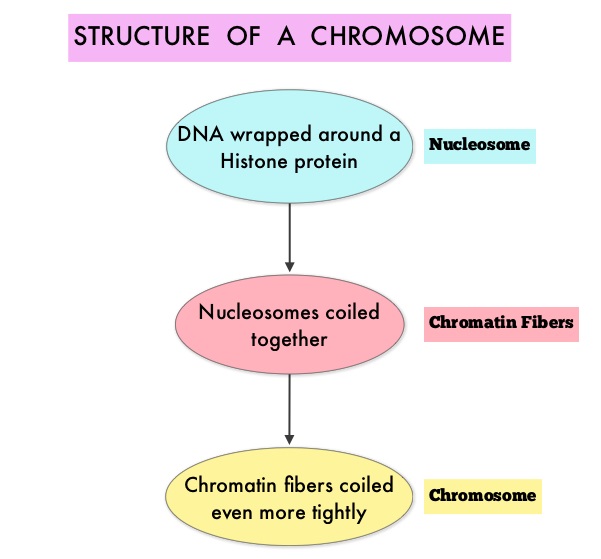 To briefly summarize the structure of a chromosome, individual loops of coiled double-helix DNA wrap around a histone protein to form a nucleosome. Nucleosomes are then coiled together to form chromatin fibers, which looks like beads on a string. The chromatin fibers are coiled even more tightly to form chromosomes. HDAC enzymes catalyze the removal of acetyl groups and regulate the level of acetylation of the histones and non-histone proteins and transcription of several genes. Hypoacetylation of histones has been associated with a condensed chromatin structure that results in the repression of gene transcription, whereas acetylated histones are associated with a more open chromatin structure and activation of gene transcription. HDACs are grouped into four major classes (Class I, II, III and IV) and regulate cell-cycle progression, cell survival, angiogenesis and immunity. The HDAC Class I enzymes are HDAC1, 2, 3 & 8 and are typically found in the nucleus where they are able to repress transcription. The HDAC Class II enzymes include HDAC4, 5, 6, 7, 9 and 10 and are able to move between the cytoplasm and nucleus and function in signal transduction. In Multiple Myeloma, the important enzyme to target is HDAC6.
To briefly summarize the structure of a chromosome, individual loops of coiled double-helix DNA wrap around a histone protein to form a nucleosome. Nucleosomes are then coiled together to form chromatin fibers, which looks like beads on a string. The chromatin fibers are coiled even more tightly to form chromosomes. HDAC enzymes catalyze the removal of acetyl groups and regulate the level of acetylation of the histones and non-histone proteins and transcription of several genes. Hypoacetylation of histones has been associated with a condensed chromatin structure that results in the repression of gene transcription, whereas acetylated histones are associated with a more open chromatin structure and activation of gene transcription. HDACs are grouped into four major classes (Class I, II, III and IV) and regulate cell-cycle progression, cell survival, angiogenesis and immunity. The HDAC Class I enzymes are HDAC1, 2, 3 & 8 and are typically found in the nucleus where they are able to repress transcription. The HDAC Class II enzymes include HDAC4, 5, 6, 7, 9 and 10 and are able to move between the cytoplasm and nucleus and function in signal transduction. In Multiple Myeloma, the important enzyme to target is HDAC6.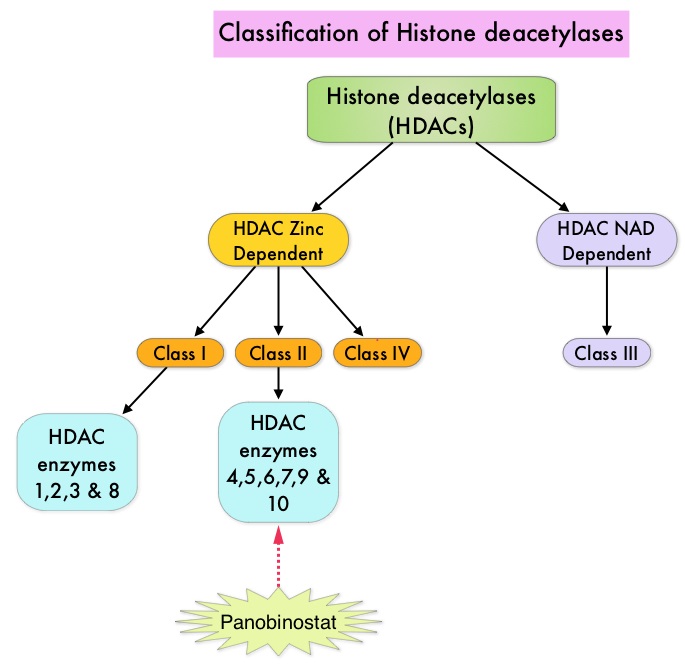 FARYDAK® is an oral, pan-histone deacetylase inhibitor which inhibits cell cycle progression and ultimately results in apoptosis. FARYDAK® inhibits the aggresome pathway of protein degradation which is upregulated when proteosome pathway is inhibited by VELCADE®. Based on preclinical data demonstrating synergy between VELCADE® and FARYDAK® in Myeloma, the PANORAMA 1 trial, enrolled patients with relapsed or refractory Multiple Myeloma who had received one to three prior lines of therapy and were not VELCADE® refractory. In this phase III trial, patients were randomly assigned to receive either FARYDAK® (N=387) or placebo (N=381), each along with IV VELCADE® and oral Dexamethasone. In this study, treatment was given in two 24 week phases. The first 24 week treatment phase was cycles 1 thru 8, where patients received placebo or FARYDAK® 20 mg orally QD 3 times a week for 2 weeks of a 3 week cycle; VELCADE® 1.3 mg/m2 IV twice weekly for 2 weeks of a 3 week cycle and Dexamethasone 20 mg PO on the day of and day after VELCADE®. Patients with clinical benefit (defined as complete response, partial response or stable disease, without significant toxicities) after the first eight cycles could proceed to the second phase of treatment in which FARYDAK® and Dexamethasone administration schedule remained the same but VELCADE® was administered once weekly for 2 weeks of the 3 week cycle. The median age was 63 years, 48% of patients had received at least two lines of therapy and 57% of patients had prior autologous stem cell transplantation and 43% had prior therapy with VELCADE®. The primary endpoint was Progression Free Survival (PFS) and secondary endpoints included Overall Survival (OS), Overall Response Rate (ORR), near Complete/Complete Response (nCR/CR) rate, Duration of Response (DOR), and safety. Among the patients enrolled in the FARYDAK® group (N = 387), 44% completed first phase of treatment and 26% completed the second phase of treatment. After a median follow up of 28 months, the primary end point of the study was met with a 37% decrease in the risk of disease progression in all the FARYDAK® group of patients compared to the placebo group (12 months vs 8.1 months, HR=0.63, P<0.0001). The median PFS was 14.65 months for those in the FARYDAK® group who completed the first phase of treatment and 17.64 months for those who completed the second phase of treatment. With regards to the secondary endpoints in the FARYDAK® vs placebo groups, the ORR was 60.7% vs 54.6% (P=0.87), nCR/CR rate was 27.6% vs 15.7% (P=0.00006), median duration of response was13.1months vs 10.9 months and median time to progression was 12.7 months vs 8.5 months respectively. It was noted that the nCR/CR rate was 52.9% for those patients who completed the second phase of treatment. The most common grade 3/4 adverse events in the FARYDAK® vs placebo arms included thrombocytopenia (67% vs 31%), neutropenia (35% vs 11%), and diarrhea (26% vs 8%) and these toxicities were manageable with dose reduction and supportive care. The authors concluded that a combination of FARYDAK®, VELCADE® and Dexamethasone significantly improves Progression Free Survival in patients with relapsed and refractory Multiple Myeloma, with manageable toxicities. Miguel JS, Hungria VTM , Yoon S, et al. 56th ASH Annual Meeting and Exposition, 2014. Abstract#4742
FARYDAK® is an oral, pan-histone deacetylase inhibitor which inhibits cell cycle progression and ultimately results in apoptosis. FARYDAK® inhibits the aggresome pathway of protein degradation which is upregulated when proteosome pathway is inhibited by VELCADE®. Based on preclinical data demonstrating synergy between VELCADE® and FARYDAK® in Myeloma, the PANORAMA 1 trial, enrolled patients with relapsed or refractory Multiple Myeloma who had received one to three prior lines of therapy and were not VELCADE® refractory. In this phase III trial, patients were randomly assigned to receive either FARYDAK® (N=387) or placebo (N=381), each along with IV VELCADE® and oral Dexamethasone. In this study, treatment was given in two 24 week phases. The first 24 week treatment phase was cycles 1 thru 8, where patients received placebo or FARYDAK® 20 mg orally QD 3 times a week for 2 weeks of a 3 week cycle; VELCADE® 1.3 mg/m2 IV twice weekly for 2 weeks of a 3 week cycle and Dexamethasone 20 mg PO on the day of and day after VELCADE®. Patients with clinical benefit (defined as complete response, partial response or stable disease, without significant toxicities) after the first eight cycles could proceed to the second phase of treatment in which FARYDAK® and Dexamethasone administration schedule remained the same but VELCADE® was administered once weekly for 2 weeks of the 3 week cycle. The median age was 63 years, 48% of patients had received at least two lines of therapy and 57% of patients had prior autologous stem cell transplantation and 43% had prior therapy with VELCADE®. The primary endpoint was Progression Free Survival (PFS) and secondary endpoints included Overall Survival (OS), Overall Response Rate (ORR), near Complete/Complete Response (nCR/CR) rate, Duration of Response (DOR), and safety. Among the patients enrolled in the FARYDAK® group (N = 387), 44% completed first phase of treatment and 26% completed the second phase of treatment. After a median follow up of 28 months, the primary end point of the study was met with a 37% decrease in the risk of disease progression in all the FARYDAK® group of patients compared to the placebo group (12 months vs 8.1 months, HR=0.63, P<0.0001). The median PFS was 14.65 months for those in the FARYDAK® group who completed the first phase of treatment and 17.64 months for those who completed the second phase of treatment. With regards to the secondary endpoints in the FARYDAK® vs placebo groups, the ORR was 60.7% vs 54.6% (P=0.87), nCR/CR rate was 27.6% vs 15.7% (P=0.00006), median duration of response was13.1months vs 10.9 months and median time to progression was 12.7 months vs 8.5 months respectively. It was noted that the nCR/CR rate was 52.9% for those patients who completed the second phase of treatment. The most common grade 3/4 adverse events in the FARYDAK® vs placebo arms included thrombocytopenia (67% vs 31%), neutropenia (35% vs 11%), and diarrhea (26% vs 8%) and these toxicities were manageable with dose reduction and supportive care. The authors concluded that a combination of FARYDAK®, VELCADE® and Dexamethasone significantly improves Progression Free Survival in patients with relapsed and refractory Multiple Myeloma, with manageable toxicities. Miguel JS, Hungria VTM , Yoon S, et al. 56th ASH Annual Meeting and Exposition, 2014. Abstract#4742
Month: February 2015
Pertuzumab, Trastuzumab, and Docetaxel in HER2-Positive Metastatic Breast Cancer
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately, 232,000 new cases of invasive breast cancer will be diagnosed in 2015 and about 40,000 women will die of the disease. The HER or erbB family of receptors consist of HER1, HER2, HER3 and HER4. Approximately 15%-20% of invasive breast cancers overexpress HER2/neu oncogene, which is a negative predictor of outcomes without systemic therapy. HERCEPTIN® (Trastuzumab) is a humanized monoclonal antibody targeting HER2. Trastuzumab binds to subdomain IV of the HER2 extracellular domain and blocks the downstream cell signaling pathways (PI3K-AKT pathway) and induces Antibody Dependent Cellular Cytotoxicity (ADCC). HERCEPTIN® in combination with chemotherapy has been proven to significantly improve Progression Free Survival and Overall Survival in patients with advanced breast cancer. Despite this benefit, majority of these patients develop progressive disease within 18 months. The tumors in these patients continue to express HER2 although the lower sensitivity to HER2 targeted agents has been attributed to HER2 independent escape mechanisms. Treatment strategies for this patient population have included switching chemotherapy in subsequent lines of treatment and continuing HERCEPTIN®, combining another HER2 targeted agent, Lapatinib (TYKERB®) with Capecitabine (XELODA®) and dual HER2 inhibition with a combination of HERCEPTIN® and TYKERB®.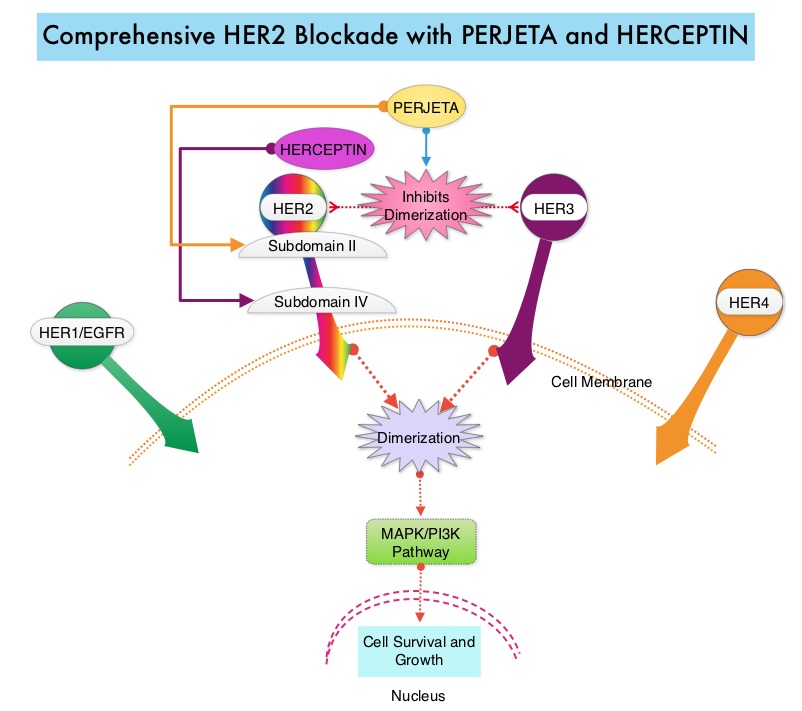 PERJETA® (Pertuzumab) is a recombinant humanized monoclonal antibody that binds to the HER2 at a different epitope of the HER2 extracellular domain (subdomain II) compared to HERCEPTIN® and prevents the dimerization of HER2 with HER3 receptor. PERJETA® stimulates antibody-dependent, cell-mediated cytotoxicity similar to HERCEPTIN®. By combining HERCEPTIN® and PERJETA®, a more comprehensive blockade of HER2 signaling can be accomplished, as these two agents bind to different HER2 epitopes and may complement each other and improve efficacy, as was demonstrated in early phase trials. The CLEOPATRA trial is a phase III study in which 808 treatment naive HER positive metastatic breast cancer patients, were randomly assigned to receive either HERCEPTIN® plus Docetaxel or this two drug combination given along with PERJETA®. PERJETA® was given as an 840 mg loading dose followed by a 420 mg maintenance dose, HERCEPTIN® was given as an 8 mg/kg loading dose followed by a 6 mg/kg maintenance dose and Docetaxel was given at 75 mg/m2 for at least 6 cycles. Treatment was administered every 3 weeks and continued until disease progression. The primary end point of this study was Progression Free Survival and secondary end points included Overall Survival, Objective Response Rate and safety. A previous analysis performed in May 2012 showed that the addition of PERJETA® to the combination of HERCEPTIN® and Docetaxel significantly prolonged Progression Free Survival compared to HERCEPTIN® plus Docetaxel alone (18.5 months vs 12.4 months) but the median overall survival had not been reached then. In this final Overall Survival analysis, at a median follow up of 50 months, median Overall Survival was 56.5 months with the PERJETA® combination compared to 40.8 months in the non-PERJETA® group (hazard ratio [HR] = 0.68; P<0.001). This meant that adding PERJETA® to HERCEPTIN® and Docetaxel increased the median Overall Survival by 15.7 months. The increase in Progression Free Survival by 6.3 months with the PERJETA® combination, was again maintained (HR = 0.68, P < 0.0001), at the time of the final analysis. The median duration of response was 20.2 months with the PERJETA® combination compared to 12.5 months in the non-PERJETA® group. The incidence of symptomatic left ventricular dysfunction as well as declines in left ventricular ejection fraction, were rare and similar between the two treatment groups. Based on the CLEOPATRA study, women with HER positive metastatic breast cancer should be considered candidates, for treatment with a combination of PERJETA®, HERCEPTIN® and Docetaxel. Swain S, Baselga J, Kim S, et al. N Engl J Med 2015; 372:724-734
PERJETA® (Pertuzumab) is a recombinant humanized monoclonal antibody that binds to the HER2 at a different epitope of the HER2 extracellular domain (subdomain II) compared to HERCEPTIN® and prevents the dimerization of HER2 with HER3 receptor. PERJETA® stimulates antibody-dependent, cell-mediated cytotoxicity similar to HERCEPTIN®. By combining HERCEPTIN® and PERJETA®, a more comprehensive blockade of HER2 signaling can be accomplished, as these two agents bind to different HER2 epitopes and may complement each other and improve efficacy, as was demonstrated in early phase trials. The CLEOPATRA trial is a phase III study in which 808 treatment naive HER positive metastatic breast cancer patients, were randomly assigned to receive either HERCEPTIN® plus Docetaxel or this two drug combination given along with PERJETA®. PERJETA® was given as an 840 mg loading dose followed by a 420 mg maintenance dose, HERCEPTIN® was given as an 8 mg/kg loading dose followed by a 6 mg/kg maintenance dose and Docetaxel was given at 75 mg/m2 for at least 6 cycles. Treatment was administered every 3 weeks and continued until disease progression. The primary end point of this study was Progression Free Survival and secondary end points included Overall Survival, Objective Response Rate and safety. A previous analysis performed in May 2012 showed that the addition of PERJETA® to the combination of HERCEPTIN® and Docetaxel significantly prolonged Progression Free Survival compared to HERCEPTIN® plus Docetaxel alone (18.5 months vs 12.4 months) but the median overall survival had not been reached then. In this final Overall Survival analysis, at a median follow up of 50 months, median Overall Survival was 56.5 months with the PERJETA® combination compared to 40.8 months in the non-PERJETA® group (hazard ratio [HR] = 0.68; P<0.001). This meant that adding PERJETA® to HERCEPTIN® and Docetaxel increased the median Overall Survival by 15.7 months. The increase in Progression Free Survival by 6.3 months with the PERJETA® combination, was again maintained (HR = 0.68, P < 0.0001), at the time of the final analysis. The median duration of response was 20.2 months with the PERJETA® combination compared to 12.5 months in the non-PERJETA® group. The incidence of symptomatic left ventricular dysfunction as well as declines in left ventricular ejection fraction, were rare and similar between the two treatment groups. Based on the CLEOPATRA study, women with HER positive metastatic breast cancer should be considered candidates, for treatment with a combination of PERJETA®, HERCEPTIN® and Docetaxel. Swain S, Baselga J, Kim S, et al. N Engl J Med 2015; 372:724-734
FARYDAK® (Panobinostat)
The FDA on February 23, 2015 granted accelerated approval to FARYDAK® for use in combination with VELCADE® (Bortezomib) and Dexamethasone for the treatment of patients with Multiple Myeloma who have received at least two prior regimens, including VELCADE® and an immunomodulatory agent. FARYDAK® capsules are a product of Novartis Pharmaceuticals.
Lenvatinib versus Placebo in Radioiodine-Refractory Thyroid Cancer
SUMMARY: The U. S. Food and Drug Administration on February 13, 2015 approved Lenvatinib (LENVIMA®) for the treatment of patients with locally recurrent or metastatic, progressive, RadioActive Iodine (RAI)-refractory Differentiated Thyroid Cancer (DTC). The American Cancer Society estimates that about 62,450 new cases of thyroid cancer will be diagnosed in the United States for 2015 and about 1,950 patients will die of the disease. Thyroid cancer is the most prevalent endocrine malignancy and is classified into three histological groups – Differentiated Thyroid Cancers (94%) which include Papillary (80%), Follicular (11%) and Hürthle cell (3%) histologies, Medullary Thyroid Carcinoma (MTC) representing 4% and Anaplastic (undifferentiated) Thyroid Carcinoma (ATC) representing about 2%. Approximately 20% of the patients with DTC develop local recurrence and 10% develop metastatic disease at 10 years following surgery, radioiodine ablation and TSH suppressive therapy. These tumors lose avidity for iodine and are considered RadioActive Iodine (RAI) refractory.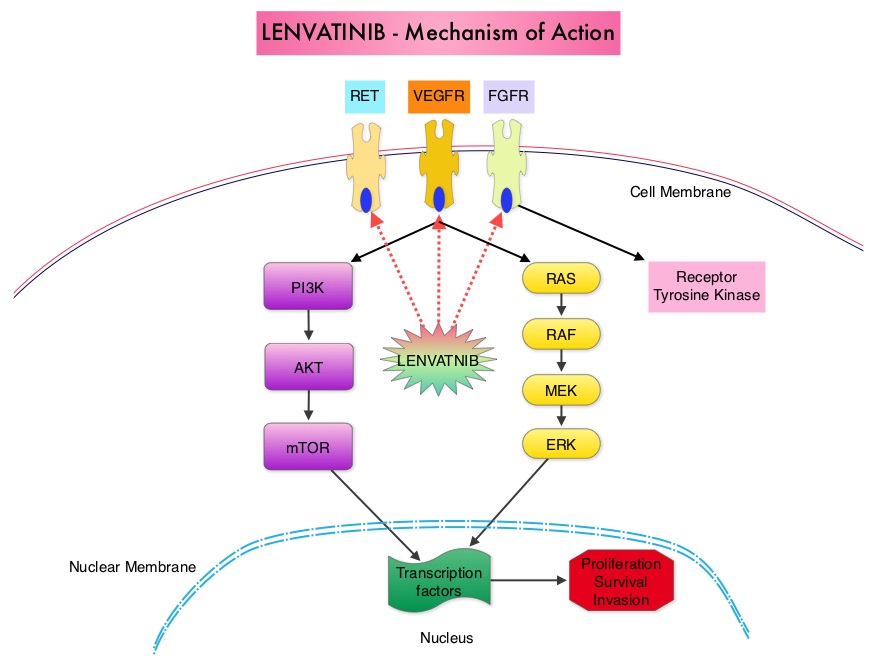 The discovery of genetic alterations in the MAP Kinase pathway as well as the PI3K (Phosphatidylinositol-3-Kinase)-AKT-mTOR pathway in thyroid tumors, has lead to the development of Tyrosine Kinase Inhibitors (TKI’s), to target these activated pathways. LENVIMA® is an oral multitargeted TKI which targets Vascular Endothelial Growth Factor Receptor (VEGFR)1-3, Fibroblast Growth factor Receptor (FGFR)1-4, Rearranged during Transfection tyrosine kinase receptor (RET), c-KIT, and Platelet Derived Growth Factor Receptor (PDGFR). LENVIMA® differs from other TKIs with antiangiogenesis properties by its ability to inhibit FGFR-1 thereby blocking the mechanisms of resistance to VEGF/VEGFR inhibitors. In addition, it controls tumor cell growth by inhibiting RET, c-KIT, and PDGFR beta and influences tumor microenvironment by inhibiting by FGFR and PDGFR beta.
The discovery of genetic alterations in the MAP Kinase pathway as well as the PI3K (Phosphatidylinositol-3-Kinase)-AKT-mTOR pathway in thyroid tumors, has lead to the development of Tyrosine Kinase Inhibitors (TKI’s), to target these activated pathways. LENVIMA® is an oral multitargeted TKI which targets Vascular Endothelial Growth Factor Receptor (VEGFR)1-3, Fibroblast Growth factor Receptor (FGFR)1-4, Rearranged during Transfection tyrosine kinase receptor (RET), c-KIT, and Platelet Derived Growth Factor Receptor (PDGFR). LENVIMA® differs from other TKIs with antiangiogenesis properties by its ability to inhibit FGFR-1 thereby blocking the mechanisms of resistance to VEGF/VEGFR inhibitors. In addition, it controls tumor cell growth by inhibiting RET, c-KIT, and PDGFR beta and influences tumor microenvironment by inhibiting by FGFR and PDGFR beta. 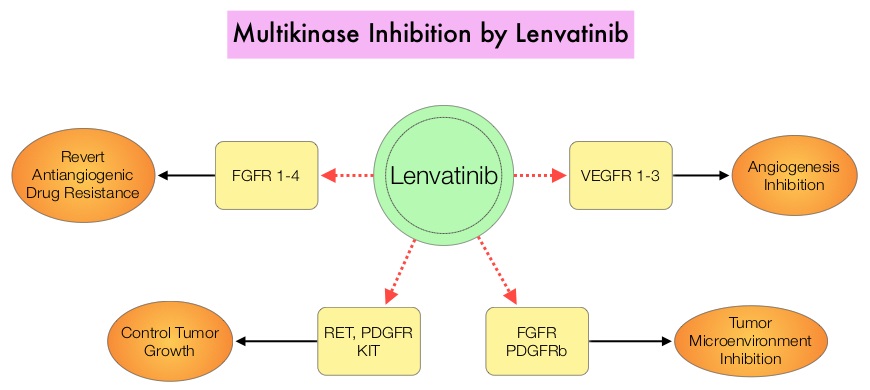 The SELECT trial is a double-blind, multicenter, phase III study in which 392 patients with advanced RAI-refractory Differentiated Thyroid Cancer (DTC) were randomly assigned in a 2:1 ratio to receive LENVIMA® 24 mg PO daily in 28-day cycles (N=261) or placebo (N=131). Both treatment groups were well balanced and pretreatment with one prior Tyrosine Kinase Inhibitor (TKI) was allowed. Patients in the placebo group were allowed to cross over and receive open-label LENVIMA®, at the time of disease progression. The primary end point was Progression Free Survival (PFS) and secondary end points included Response Rate, Overall Survival, and safety. The median Progression Free Survival was 18.3 months in the LENVIMA® group and 3.6 months in the placebo group (HR= 0.21; P<0.001). This benefit in PFS associated with LENVIMA® was observed in all pre-specified subgroup of patients including those who had received one prior treatment with a TKI. The objective response rate with LENVIMA® was 64.8% versus 1.5% with placebo (P<0.001) and the median overall survival was not reached in either group. The most frequently reported grade 3 or more adverse events in the LENVIMA® group included hypertension (42.9%) and proteinuria (10%). Approximately 14% of the patients in the LENVIMA® group discontinued the drug due to adverse effects. Exploratory biomarker analyses were performed for BRAF and RAS mutations on tumor tissue and it was noted that LENVIMA® benefitted patients regardless of BRAFor RAS mutation status. The authors concluded that LENVIMA® decreased the risk of disease progression by 79% as compared with placebo and was associated with significant improvements in objective Response Rate among patients with RAI-refractory thyroid cancer. NEXAVAR® (Sorafenib), another multitargeted TKI, is presently available for this group of patients and therefore proper sequencing of LENVIMA® and NEXAVAR® remains unknown although it appears that LENVIMA® has a markedly higher Progression Free Survival compared to NEXAVAR®. Schlumberger M, Tahara M, Wirth LJ, et al. N Engl J Med 2015; 372:621-630
The SELECT trial is a double-blind, multicenter, phase III study in which 392 patients with advanced RAI-refractory Differentiated Thyroid Cancer (DTC) were randomly assigned in a 2:1 ratio to receive LENVIMA® 24 mg PO daily in 28-day cycles (N=261) or placebo (N=131). Both treatment groups were well balanced and pretreatment with one prior Tyrosine Kinase Inhibitor (TKI) was allowed. Patients in the placebo group were allowed to cross over and receive open-label LENVIMA®, at the time of disease progression. The primary end point was Progression Free Survival (PFS) and secondary end points included Response Rate, Overall Survival, and safety. The median Progression Free Survival was 18.3 months in the LENVIMA® group and 3.6 months in the placebo group (HR= 0.21; P<0.001). This benefit in PFS associated with LENVIMA® was observed in all pre-specified subgroup of patients including those who had received one prior treatment with a TKI. The objective response rate with LENVIMA® was 64.8% versus 1.5% with placebo (P<0.001) and the median overall survival was not reached in either group. The most frequently reported grade 3 or more adverse events in the LENVIMA® group included hypertension (42.9%) and proteinuria (10%). Approximately 14% of the patients in the LENVIMA® group discontinued the drug due to adverse effects. Exploratory biomarker analyses were performed for BRAF and RAS mutations on tumor tissue and it was noted that LENVIMA® benefitted patients regardless of BRAFor RAS mutation status. The authors concluded that LENVIMA® decreased the risk of disease progression by 79% as compared with placebo and was associated with significant improvements in objective Response Rate among patients with RAI-refractory thyroid cancer. NEXAVAR® (Sorafenib), another multitargeted TKI, is presently available for this group of patients and therefore proper sequencing of LENVIMA® and NEXAVAR® remains unknown although it appears that LENVIMA® has a markedly higher Progression Free Survival compared to NEXAVAR®. Schlumberger M, Tahara M, Wirth LJ, et al. N Engl J Med 2015; 372:621-630
A Prospective Multicenter Study Of The Bruton’s Tyrosine Kinase Inhibitor Ibrutinib In Patients With Relapsed Or Refractory Waldenstrom’s Macroglobulinemia
SUMMARY: The US Food and Drug Administration on January 29, 2015, approved Ibrutinib (IMBRUVICA®) for the treatment of patients with Waldenstrom Macroglobulinemia (WM). Waldenstrom Macroglobulinemia is classified as a LymphoPlasmacytic Lymphoma (LPL) according to the Revised European American Lymphoma (REAL) and WHO (World Health Organization) classifications. It is estimated that about 1500 new cases of WM will be diagnosed each year in the US. Chromosome 6q deletion detected by FISH technique on bone marrow evaluation, is seen in 40-50% of the patients with WM. Whole genome sequencing of lymphoplasmacytic cells has revealed activating somatic mutations in MYD88 (L265P) and CXCR4 genes, in Waldenstrom Macroglobulinemia (WM).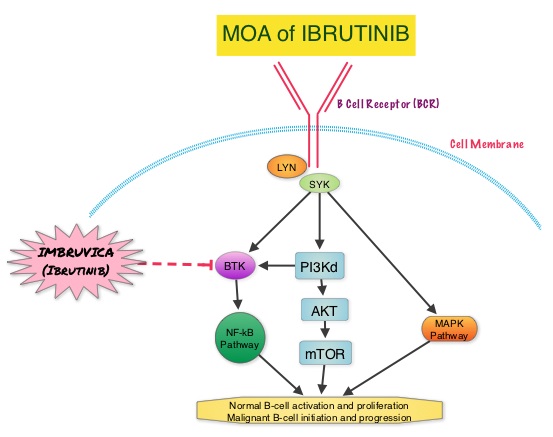 About 90% of patients with Waldenstrom Macroglobulinemia demonstrate a mutation in chromosome 3p (MYD88 L265P), which is specific to WM and may be an early oncogenic event in WM pathogenesis.. It appears that MYD88 L265P promotes malignant cell proliferation via the Bruton’s Tyrosine Kinase (BTK) signaling pathway. Mutations in CXCR4 gene are present in 30% of patients with WM, and their expression induces BTK activity and may confer resistance to BTK inhibitors. IMBRUVICA® (Ibrutinib) is an oral, irreversible inhibitor of BTK and inhibits cell proliferation and promotes programmed cell death (Apoptosis). Preliminary studies in WM patients have revealed that IMBRUVICA® prevents the binding of MYD88 L265P (mutated gene) to BTK thereby selectively killing tumor cells. On the other hand it was noted that a major response to IMBRUVICA® was less likely when mutations in CXCR4 gene were present in the tumor cells. With this molecular understanding of WM, the authors enrolled 63 patients with relapsed/refractory symptomatic WM and were treated with IMBRUVICA®, 420 mg PO daily for 2 years or until disease progression or unacceptable toxicity. Anemia was main indication for treatment initiation (87.3% of patients) and the mean baseline hemoglobin level was 10.5 g/dL, mean serum IgM level was 3610 mg/dL, 70% had bone marrow involvement and 60% of patients had lymphadenopathy.
About 90% of patients with Waldenstrom Macroglobulinemia demonstrate a mutation in chromosome 3p (MYD88 L265P), which is specific to WM and may be an early oncogenic event in WM pathogenesis.. It appears that MYD88 L265P promotes malignant cell proliferation via the Bruton’s Tyrosine Kinase (BTK) signaling pathway. Mutations in CXCR4 gene are present in 30% of patients with WM, and their expression induces BTK activity and may confer resistance to BTK inhibitors. IMBRUVICA® (Ibrutinib) is an oral, irreversible inhibitor of BTK and inhibits cell proliferation and promotes programmed cell death (Apoptosis). Preliminary studies in WM patients have revealed that IMBRUVICA® prevents the binding of MYD88 L265P (mutated gene) to BTK thereby selectively killing tumor cells. On the other hand it was noted that a major response to IMBRUVICA® was less likely when mutations in CXCR4 gene were present in the tumor cells. With this molecular understanding of WM, the authors enrolled 63 patients with relapsed/refractory symptomatic WM and were treated with IMBRUVICA®, 420 mg PO daily for 2 years or until disease progression or unacceptable toxicity. Anemia was main indication for treatment initiation (87.3% of patients) and the mean baseline hemoglobin level was 10.5 g/dL, mean serum IgM level was 3610 mg/dL, 70% had bone marrow involvement and 60% of patients had lymphadenopathy. 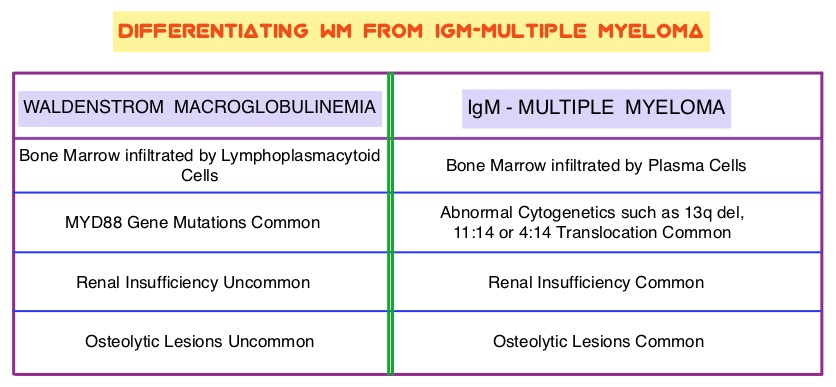 Sanger sequencing was used to determine MYD88 and CXCR4 mutations in the bone marrow lymphoplasmacytic cells. At best response, the median serum IgM levels declined to 1340 mg/dL (P<0.00001), median hemoglobin rose to 12.6 g/dL, (P<0.00001). At 6 months, bone marrow assessment post treatment, demonstrated a reduction in WM disease involvement from 70% to 45% (P=0.0006). With a median follow up at 6 cycles, the best overall response rate was 81% and a median time to response was 4 weeks. In patients who underwent tumor sequencing, mutations in CXCR4 gene impacted response rates. The major response rate for patients with wild-type CXCR4 gene was 77% compares to 30% in those with CXCR4 mutations (p=0.018). Further, patients with wild-type CXCR4 also had increased peripheral lymphocytosis following treatment with IMBRUVICA® compared to those with CXCR4 mutations (P=0.001). The most common more than grade 2 treatment related toxicities included thrombocytopenia (14.3%) and neutropenia (19.1%). The authors concluded that IMBRUVICA® is highly active in patients with relapsed or refractory Waldenstrom Macroglobulinemia, with rapid reductions in serum IgM level and improved hemoglobin levels. The presence of CXCR4 mutations negatively impact response rates in this patient group. Treon SP, Tripsas CK, Yang G, et al. Presented at: 55th ASH Annual Meeting; December 7-10, 2013; New Orleans, LA. Abstract 251.
Sanger sequencing was used to determine MYD88 and CXCR4 mutations in the bone marrow lymphoplasmacytic cells. At best response, the median serum IgM levels declined to 1340 mg/dL (P<0.00001), median hemoglobin rose to 12.6 g/dL, (P<0.00001). At 6 months, bone marrow assessment post treatment, demonstrated a reduction in WM disease involvement from 70% to 45% (P=0.0006). With a median follow up at 6 cycles, the best overall response rate was 81% and a median time to response was 4 weeks. In patients who underwent tumor sequencing, mutations in CXCR4 gene impacted response rates. The major response rate for patients with wild-type CXCR4 gene was 77% compares to 30% in those with CXCR4 mutations (p=0.018). Further, patients with wild-type CXCR4 also had increased peripheral lymphocytosis following treatment with IMBRUVICA® compared to those with CXCR4 mutations (P=0.001). The most common more than grade 2 treatment related toxicities included thrombocytopenia (14.3%) and neutropenia (19.1%). The authors concluded that IMBRUVICA® is highly active in patients with relapsed or refractory Waldenstrom Macroglobulinemia, with rapid reductions in serum IgM level and improved hemoglobin levels. The presence of CXCR4 mutations negatively impact response rates in this patient group. Treon SP, Tripsas CK, Yang G, et al. Presented at: 55th ASH Annual Meeting; December 7-10, 2013; New Orleans, LA. Abstract 251.
Lenvatinib versus Placebo in Radioiodine-Refractory Thyroid Cancer
SUMMARY: The U. S. Food and Drug Administration on February 13, 2015 approved Lenvatinib (LENVIMA®) for the treatment of patients with locally recurrent or metastatic, progressive, RadioActive Iodine (RAI)-refractory Differentiated Thyroid Cancer (DTC). The American Cancer Society estimates that about 62,450 new cases of thyroid cancer will be diagnosed in the United States for 2015 and about 1,950 patients will die of the disease. Thyroid cancer is the most prevalent endocrine malignancy and is classified into three histological groups – Differentiated Thyroid Cancers (94%) which include Papillary (80%), Follicular (11%) and Hürthle cell (3%) histologies, Medullary Thyroid Carcinoma (MTC) representing 4% and Anaplastic (undifferentiated) Thyroid Carcinoma (ATC) representing about 2%. Approximately 20% of the patients with DTC develop local recurrence and 10% develop metastatic disease at 10 years following surgery, radioiodine ablation and TSH suppressive therapy. These tumors lose avidity for iodine and are considered RadioActive Iodine (RAI) refractory. The discovery of genetic alterations in the MAP Kinase pathway as well as the PI3K (Phosphatidylinositol-3-Kinase)-AKT-mTOR pathway in thyroid tumors, has lead to the development of Tyrosine Kinase Inhibitors (TKI’s), to target these activated pathways. LENVIMA® is an oral multitargeted TKI which targets Vascular Endothelial Growth Factor Receptor (VEGFR)1-3, Fibroblast Growth factor Receptor (FGFR)1-4, Rearranged during Transfection tyrosine kinase receptor (RET), c-KIT, and Platelet Derived Growth Factor Receptor (PDGFR). LENVIMA® differs from other TKIs with antiangiogenesis properties by its ability to inhibit FGFR-1 thereby blocking the mechanisms of resistance to VEGF/VEGFR inhibitors. In addition, it controls tumor cell growth by inhibiting RET, c-KIT, and PDGFR beta and influences tumor microenvironment by inhibiting by FGFR and PDGFR beta. The SELECT trial is a double-blind, multicenter, phase III study in which 392 patients with advanced RAI-refractory Differentiated Thyroid Cancer (DTC) were randomly assigned in a 2:1 ratio to receive LENVIMA® 24 mg PO daily in 28-day cycles (N=261) or placebo (N=131). Both treatment groups were well balanced and pretreatment with one prior Tyrosine Kinase Inhibitor (TKI) was allowed. Patients in the placebo group were allowed to cross over and receive open-label LENVIMA®, at the time of disease progression. The primary end point was Progression Free Survival (PFS) and secondary end points included Response Rate, Overall Survival, and safety. The median Progression Free Survival was 18.3 months in the LENVIMA® group and 3.6 months in the placebo group (HR= 0.21; P<0.001). This benefit in PFS associated with LENVIMA® was observed in all pre-specified subgroup of patients including those who had received one prior treatment with a TKI. The objective response rate with LENVIMA® was 64.8% versus 1.5% with placebo (P<0.001) and the median overall survival was not reached in either group. The most frequently reported grade 3 or more adverse events in the LENVIMA® group included hypertension (42.9%) and proteinuria (10%). Approximately 14% of the patients in the LENVIMA® group discontinued the drug due to adverse effects. Exploratory biomarker analyses were performed for BRAF and RAS mutations on tumor tissue and it was noted that LENVIMA® benefitted patients regardless of BRAFor RAS mutation status. The authors concluded that LENVIMA® decreased the risk of disease progression by 79% as compared with placebo and was associated with significant improvements in objective Response Rate among patients with RAI-refractory thyroid cancer. NEXAVAR® (Sorafenib), another multitargeted TKI, is presently available for this group of patients and therefore proper sequencing of LENVIMA® and NEXAVAR® remains unknown although it appears that LENVIMA® has a markedly higher Progression Free Survival compared to NEXAVAR®. Schlumberger M, Tahara M, Wirth LJ, et al. N Engl J Med 2015; 372:621-630
IBRANCE® (Palbociclib)
The FDA on February 3, 2015 granted accelerated approval to IBRANCE® for use in combination with FEMARA® (Letrozole) for the treatment of postmenopausal women with Estrogen Receptor (ER) positive, human epidermal growth factor receptor 2 (HER2) negative advanced Breast Cancer, as initial endocrine-based therapy for their metastatic disease. IBRANCE® is a product of Pfizer, Inc.
Platelet Transfusion A Clinical Practice Guideline from the AABB
SUMMARY: Platelets are often transfused preemptively to reduce the risk for spontaneous bleeding in patients who are thrombocytopenic following chemotherapy or hematopoietic stem cell transplantation. In the United States, a little over 2 million platelet units are transfused annually. The risks associated with platelet transfusion such as febrile and allergic reactions, Transfusion Related Acute Lung Injury and infections, have to be taken into consideration before transfusion is planned. Further, unlike other blood products, platelets must be stored at room temperature and this limits the platelet unit shelf life to only 5 days, to prevent the risk for bacterial growth during storage. This in turn is an additional burden to the hospital blood banks. The AABB (American Association of Blood Banks) commissioned and funded the development of platelet transfusion guidelines with the help of 21 experts from various specialties of medicine, after a systematic review of 17 randomized controlled trials and 53 observational studies. A platelet unit in this guideline refers to 1 apheresis platelet unit or a pool of 4-6 whole blood derived platelet concentrates, containing approximately 3- 4 x 1011 platelets. Thrombocytopenia refers to a platelet count below the lower limit of the normal range used by the laboratory performing the count. Six recommendations were made for 4 different clinical settings.
Hospitalized Adult Patients with Therapy-Induced Hypoproliferative Thrombocytopenia
Recommendation 1: The AABB recommends that platelets should be transfused prophylactically to reduce the risk for spontaneous bleeding in hospitalized adult patients with therapy-induced hypoproliferative thrombocytopenia. The AABB recommends transfusing hospitalized adult patients with a platelet count of 10 × 109 cells/L or less to reduce the risk for spontaneous bleeding. The AABB recommends transfusing up to a single apheresis unit or equivalent. Greater doses are not more effective, and lower doses equal to one half of a standard apheresis unit are equally effective.
Adult Patients Having Minor Invasive Procedures
Recommendation 2: The AABB suggests prophylactic platelet transfusion for patients having elective central venous catheter placement with a platelet count less than 20 × 109 cells/L.
Recommendation 3: The AABB suggests prophylactic platelet transfusion for patients having elective diagnostic lumbar puncture with a platelet count less than 50 × 109 cells/L.
Adult Patients Having Major Elective Non-neuraxial Surgery
Recommendation 4: The AABB suggests prophylactic platelet transfusion for patients having major elective non-neuraxial surgery with a platelet count less than 50 × 109 cells/L.
Recommendation 5: The AABB recommends against routine prophylactic platelet transfusion for patients who are non-thrombocytopenic and have cardiac surgery with cardiopulmonary bypass. The AABB suggests platelet transfusion for patients having bypass who exhibit perioperative bleeding with thrombocytopenia and/or evidence of platelet dysfunction.
Adult Patients Receiving Antiplatelet Therapy Who Have Intracranial Hemorrhage (Traumatic or Spontaneous)
Recommendation 6: The AABB cannot recommend for or against platelet transfusion for patients receiving antiplatelet therapy who have intracranial hemorrhage (traumatic or spontaneous).
The authors acknowledge that the platelet transfusion threshold recommendations made by the various medical specialty societies, may be different, but the evidence based recommendations provided herein, will complement clinical judgment, as individualized decisions are made, to transfuse platelets. Kaufman RM, Djulbegovic B, Gernsheimer T, et al. Ann Intern Med. 2015;162:205-213.
The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18) a randomised phase 2 study
SUMMARY:The U. S. Food and Drug Administration on February 3, 2015 granted accelerated approval to IBRANCE® (Palbociclib), for use in combination with FEMARA® (Letrozole), for the treatment of postmenopausal women with Estrogen Receptor (ER)-positive, Human Epidermal growth factor Receptor 2 (HER2) negative advanced Breast Cancer, as initial endocrine-based therapy for their metastatic disease. Approximately 60% of the breast tumors express Estrogen Receptors and these patients are often treated with anti-estrogen therapy as first line treatment.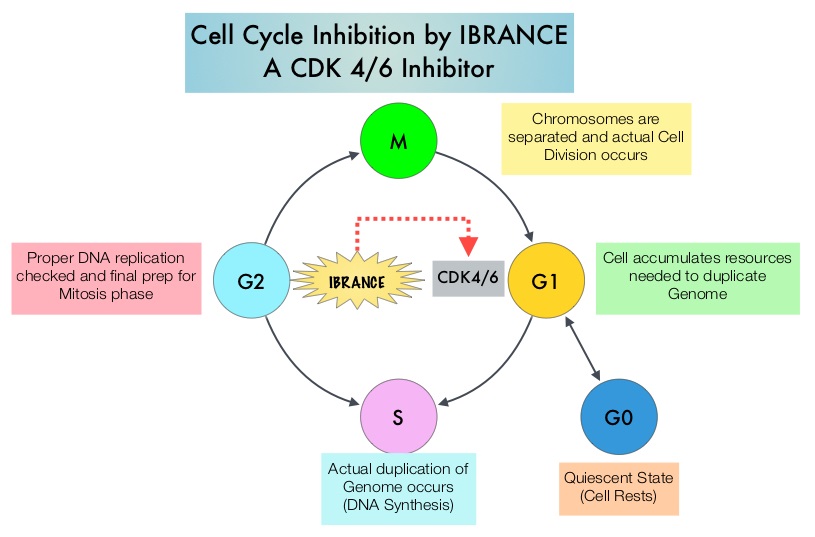 Cyclin Dependent Kinases (CDK) play a very important role to facilitate orderly and controlled progression of the cell cycle. Genetic alterations in these kinases and their regulatory proteins have been implicated in various malignancies. IBRANCE® is a reversible, oral, small molecule inhibitor of Cyclin Dependent Kinases (CDK) 4/6 and is the first CDK inhibitor approved by the FDA. This agent in pre-clinical studies was noted to reduce cellular proliferation of Estrogen Receptor positive Breast Cancer cell lines by blocking progression of cells from G1 into S phase of the cell cycle and was also noted to be synergistic with anti-estrogens. The approval of IBRANCE® was based on an open-label, randomized, multicenter, phase II study which included postmenopausal women with ER-positive, HER2-negative, advanced (locally advanced or metastatic) Breast Cancer who had not received previous systemic treatment for advanced disease. This trial enrolled and randomly assigned 165 patients, to receive either IBRANCE® 125 mg PO daily for 21 consecutive days, followed by 7 days off treatment) plus FEMARA® (2.5 mg PO daily continuously throughout the 28-day cycle (N=84) or FEMARA® alone (N=81). Among the 165 patients, 43% had received chemotherapy and 33% had received anti-hormonal therapy as a neoadjuvant or adjuvant treatment. Fortynine percent (49%) of patients had no prior systemic therapy in the neoadjuvant or adjuvant setting. The majority of patients (98%) had stage IV disease, 48% had visceral disease and 17% had bone only disease. The primary endpoint was investigator-assessed Progression Free Survival. The median PFS was 20.2 months in the IBRANCE® plus FEMARA® group and 10.2 months in the FEMARA® alone group (HR=0.488, P=0•0004). In patients with measurable disease, the Overall Response rate was higher in the IBRANCE® plus FEMARA® group compared to the FEMARA® alone group (55% versus 39%, P=0.04). The median duration of response for those who had a partial or complete response was 20.3 months with a combination of IBRANCE® plus FEMARA® vs 11.1 months with FEMARA® alone. The most common Grade 3–4 adverse events in the IBRANCE® plus FEMARA® group were neutropenia and leucopenia but without any cases of neutropenic fever. Unlike chemotherapy, the duration of neutropenia was brief with rapid recovery of blood counts. The authors concluded that the addition of IBRANCE® to FEMARA® significantly improved Progression Free Survival in women with advanced Estrogen Receptor positive and HER2-negative breast cancer. This combination may therefore be a reasonable treatment option in this patient group, soon after metastatic disease is diagnosed. Biomarkers expression (cyclinD1 amplification and/or loss of p16) had no impact on outcomes suggesting that the biomarker for IBRANCE® may be the Estrogen Receptor itself, rather than CDK4/6 kinases. Finn RS, Crown JP, Lang I, et al. Lancet Oncol 2015; 16:25-35
Cyclin Dependent Kinases (CDK) play a very important role to facilitate orderly and controlled progression of the cell cycle. Genetic alterations in these kinases and their regulatory proteins have been implicated in various malignancies. IBRANCE® is a reversible, oral, small molecule inhibitor of Cyclin Dependent Kinases (CDK) 4/6 and is the first CDK inhibitor approved by the FDA. This agent in pre-clinical studies was noted to reduce cellular proliferation of Estrogen Receptor positive Breast Cancer cell lines by blocking progression of cells from G1 into S phase of the cell cycle and was also noted to be synergistic with anti-estrogens. The approval of IBRANCE® was based on an open-label, randomized, multicenter, phase II study which included postmenopausal women with ER-positive, HER2-negative, advanced (locally advanced or metastatic) Breast Cancer who had not received previous systemic treatment for advanced disease. This trial enrolled and randomly assigned 165 patients, to receive either IBRANCE® 125 mg PO daily for 21 consecutive days, followed by 7 days off treatment) plus FEMARA® (2.5 mg PO daily continuously throughout the 28-day cycle (N=84) or FEMARA® alone (N=81). Among the 165 patients, 43% had received chemotherapy and 33% had received anti-hormonal therapy as a neoadjuvant or adjuvant treatment. Fortynine percent (49%) of patients had no prior systemic therapy in the neoadjuvant or adjuvant setting. The majority of patients (98%) had stage IV disease, 48% had visceral disease and 17% had bone only disease. The primary endpoint was investigator-assessed Progression Free Survival. The median PFS was 20.2 months in the IBRANCE® plus FEMARA® group and 10.2 months in the FEMARA® alone group (HR=0.488, P=0•0004). In patients with measurable disease, the Overall Response rate was higher in the IBRANCE® plus FEMARA® group compared to the FEMARA® alone group (55% versus 39%, P=0.04). The median duration of response for those who had a partial or complete response was 20.3 months with a combination of IBRANCE® plus FEMARA® vs 11.1 months with FEMARA® alone. The most common Grade 3–4 adverse events in the IBRANCE® plus FEMARA® group were neutropenia and leucopenia but without any cases of neutropenic fever. Unlike chemotherapy, the duration of neutropenia was brief with rapid recovery of blood counts. The authors concluded that the addition of IBRANCE® to FEMARA® significantly improved Progression Free Survival in women with advanced Estrogen Receptor positive and HER2-negative breast cancer. This combination may therefore be a reasonable treatment option in this patient group, soon after metastatic disease is diagnosed. Biomarkers expression (cyclinD1 amplification and/or loss of p16) had no impact on outcomes suggesting that the biomarker for IBRANCE® may be the Estrogen Receptor itself, rather than CDK4/6 kinases. Finn RS, Crown JP, Lang I, et al. Lancet Oncol 2015; 16:25-35
Pre- and Post-diagnosis Physical Activity, Television Viewing, and Mortality Among Patients With Colorectal Cancer in the National Institutes of Health–AARP Diet and Health Study
SUMMARY:The American Cancer Society estimates that approximately 137,000 new cases of colorectal cancer were diagnosed in the United States in 2014 and over 50,000 died of the disease. Even though colon cancer localized to the bowel is potentially curable with surgery and adjuvant chemotherapy, advanced colon cancer is often incurable. Standard chemotherapy when combined with anti EGFR (Epidermal Growth Factor Receptor) targeted monoclonal antibodies such as VECTIBIX® (Panitumumab) and ERBITUX® (Cetuximab) as well as anti VEGF agent AVASTIN® (Bevacizumab), have demonstrated improvement in Progression Free Survival and Overall Survival. Reducing risk of death and improving survival even further with leisure time physical activity is the topic of this discussion. The most prevalent sedentary behavior, watching TV, has been associated with poorer survival in the general population. Several studies have suggested protective effects of physical activity on cancer recurrence although this has not been conclusive. Pooled data from other studies have shown that lack of physical activity among survivors of ColoRectal Cancer (CRC) has been associated with higher mortality risk. However, the independent effects of physical activity before and after colorectal cancer diagnosis, has remained unclear and the association between watching TV and mortality in survivors of CRC has not been defined.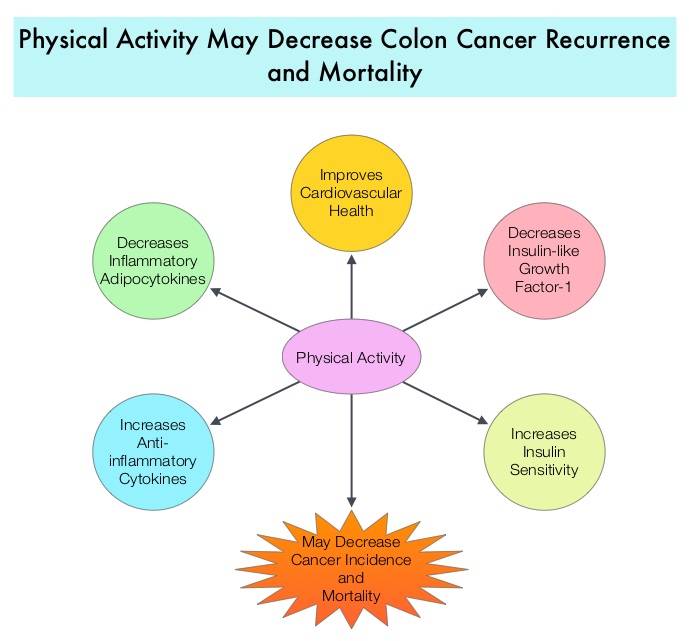 There are several biologic mechanisms that explain the association between sedentary life style, physical activity, and mortality. The adipose tissue in the body in addition to serving as the storage site for energy also releases adipokines, which have pro-inflammatory and anti-inflammatory properties. Physical activity decreases inflammatory adipocytokines and increases anti-inflammatory cytokines and thereby could affect cancer incidence and mortality. Also, physical activity has been shown to increase insulin sensitivity. Several studies have shown increased risk of CRC, increased angiogenesis, tumor growth, and anti-apoptotic activity with higher circulating insulin and insulin-like growth factor-1 and lower insulin-binding protein levels. Physical activity also improves cardiovascular health by lowering blood pressure. The authors in this study explored the impact of lifestyle such as moderate to vigorous intensity physical activity level and TV viewing time (sedentary life style) on ColoRectal Cancer mortality among CRC survivors, with particular attention to pre and post cancer diagnosis contributing factors. Data was collected from a large cohort of patients enrolled in the National Institutes of Health Diet and Health Study and the associations were analyzed between pre CRC diagnosis (N=3797) and post CRC diagnosis (N= 1759) life styles (physical activity and TV watching) and overall and disease-specific mortality. It was noted that patients who had 7 or more hours of weekly leisure physical activity before their diagnosis of CRC had a 20% lower risk of all-cause mortality compared to those who had a sedentary life style and were not engaged in any leisure time activity (HR=0.80; P=0.02). Patients who were engaged in 7 or more hours of weekly leisure physical activity post CRC diagnosis had a 31% lower risk of all-cause mortality compared to those who reported no activity (HR=0.69; P=0.006). This benefit was noted independent of pre-diagnosis activity. Amongst those who reported sedentary lifestyle, patients who spent 5 or more TV hours per day before CRC diagnosis had a 22% increased risk in all-cause mortality compared to those who watched no more than 2 hours of TV per day (HR=1.22; P=0.002). A similar trend of increased risk of all-cause mortality was seen in those who spent more TV hours, post diagnosis although this was not statistically significant. The authors concluded that in patients with CRC, leisure time physical activity was inversely associated with all-cause mortality, whereas more TV hours was associated with increased mortality risk. Health Care providers should therefore proactively promote increasing physical activity and minimizing TV hours, among survivors of ColoRectal Cancer. Arem H, Pfeiffer RM, Engels EA, et al. J Clin Oncol 2015; 33:180-188
There are several biologic mechanisms that explain the association between sedentary life style, physical activity, and mortality. The adipose tissue in the body in addition to serving as the storage site for energy also releases adipokines, which have pro-inflammatory and anti-inflammatory properties. Physical activity decreases inflammatory adipocytokines and increases anti-inflammatory cytokines and thereby could affect cancer incidence and mortality. Also, physical activity has been shown to increase insulin sensitivity. Several studies have shown increased risk of CRC, increased angiogenesis, tumor growth, and anti-apoptotic activity with higher circulating insulin and insulin-like growth factor-1 and lower insulin-binding protein levels. Physical activity also improves cardiovascular health by lowering blood pressure. The authors in this study explored the impact of lifestyle such as moderate to vigorous intensity physical activity level and TV viewing time (sedentary life style) on ColoRectal Cancer mortality among CRC survivors, with particular attention to pre and post cancer diagnosis contributing factors. Data was collected from a large cohort of patients enrolled in the National Institutes of Health Diet and Health Study and the associations were analyzed between pre CRC diagnosis (N=3797) and post CRC diagnosis (N= 1759) life styles (physical activity and TV watching) and overall and disease-specific mortality. It was noted that patients who had 7 or more hours of weekly leisure physical activity before their diagnosis of CRC had a 20% lower risk of all-cause mortality compared to those who had a sedentary life style and were not engaged in any leisure time activity (HR=0.80; P=0.02). Patients who were engaged in 7 or more hours of weekly leisure physical activity post CRC diagnosis had a 31% lower risk of all-cause mortality compared to those who reported no activity (HR=0.69; P=0.006). This benefit was noted independent of pre-diagnosis activity. Amongst those who reported sedentary lifestyle, patients who spent 5 or more TV hours per day before CRC diagnosis had a 22% increased risk in all-cause mortality compared to those who watched no more than 2 hours of TV per day (HR=1.22; P=0.002). A similar trend of increased risk of all-cause mortality was seen in those who spent more TV hours, post diagnosis although this was not statistically significant. The authors concluded that in patients with CRC, leisure time physical activity was inversely associated with all-cause mortality, whereas more TV hours was associated with increased mortality risk. Health Care providers should therefore proactively promote increasing physical activity and minimizing TV hours, among survivors of ColoRectal Cancer. Arem H, Pfeiffer RM, Engels EA, et al. J Clin Oncol 2015; 33:180-188