
SUMMARY: The American Cancer Society estimates that about 71,850 people will be diagnosed with Non-Hodgkin Lymphoma (NHL) in the United States and about 19,800 individuals will die of this disease. Approximately 20% of all NHLs are Follicular Lymphomas. Follicular Lymphoma is the most indolent form and second most common form of all NHLs and they are a heterogeneous group of lymphoproliferative malignancies. Advanced stage Follicular Lymphomas are not curable and as such prolonging Progression Free Survival (PFS) and Overall Survival (OS) while maintaining quality of life (QoL), has been the goals of treatment intervention. Asymptomatic patients with FL are generally considered candidates for “watch and wait” approach, whereas those with B symptoms (fever, night sweats, and weight loss), painful lymphadenopathy/splenomegaly, organ compromise and cytopenias are generally considered candidates for therapy. Follicular Lymphoma International Prognostic Index (FLIPI) is of prognostic value and is used to help with treatment choices. Several studies have been underway evaluating the association between serum Vitamin D levels and cancer. Previously published studies have shown a relationship between Vitamin D deficiency and poor outcomes in patients with Diffuse Large B Cell Lymphoma and Chronic Lymphocytic Leukemia. The beneficial effects of Vitamin D in malignancies has been attributed to its antiproliferative and antiangiogenic properties, as well as its effects on cell differentiation, promotion of apoptosis and its ability to decreases oxidative DNA damage. Further, macrophages play an important role in the human body’s response to therapy with monoclonal antibodies, an integral part of Follicular Lymphoma therapies and low serum Vitamin D levels may interfere with macrophage function and this may explain poor outcomes in some Follicular Lymphoma patients with low Vitamin D levels
The beneficial effects of Vitamin D in malignancies has been attributed to its antiproliferative and antiangiogenic properties, as well as its effects on cell differentiation, promotion of apoptosis and its ability to decreases oxidative DNA damage. Further, macrophages play an important role in the human body’s response to therapy with monoclonal antibodies, an integral part of Follicular Lymphoma therapies and low serum Vitamin D levels may interfere with macrophage function and this may explain poor outcomes in some Follicular Lymphoma patients with low Vitamin D levels
Month: August 2015
XTANDI® After TAXOTERE® and ZYTIGA® Treatment in Prostate Cancer
SUMMARY: Prostate cancer is the most common cancer in American men with the exclusion of skin cancer and 1 in 7 men will be diagnosed with prostate cancer during their lifetime. It is estimated that in the United States, about 220,800 new cases of prostate cancer will be diagnosed in 2015 and over 27,000 men will die of the disease. The development and progression of prostate cancer is driven by androgens. Androgen Deprivation Therapy (ADT) has therefore been the cornerstone of treatment of advanced prostate cancer and is the first treatment intervention for hormone sensitive prostate cancer. Chemotherapy is usually considered for patients who progress on hormone therapy (Castrate Resistant Prostate Cancer-CRPC) and TAXOTERE® (Docetaxel) has been shown to improve Overall Survival (OS) of metastatic prostate cancer patients, who had progressed on Androgen Deprivation Therapy. Tumors in patients with CRPC are not androgen independent and continue to rely on Androgen Receptor signaling and two oral agents are presently available for metastatic CRPC. They include ZYTIGA® (Abiraterone) and XTANDI® (Enzalutamide). ZYTIGA® inhibits CYP 17A1 enzyme thus decreasing androgen biosynthesis and depletes adrenal and intratumoral androgens. XTANDI® competes with Testosterone and Dihydrotestosterone and avidly binds to the Androgen Receptor (AR), thereby inhibiting AR signaling and in addition inhibits translocation of the AR into the nucleus and thus inhibits the transcriptional activities of the AR. There is presently very little guidance with regards to the sequencing of these two oral agents after progression on TAXOTERE®, in patients with metastatic CRPC. ZYTIGA® was approved initially by the FDA in April 2011, for use in combination with prednisone for the treatment of patients with metastatic CRPC, who had received prior chemotherapy containing TAXOTERE®. Treatment with ZYTIGA® resulted in a 35% reduction in the risk of death and a 36% increase in median Overall Survival (OS) compared with placebo. Subsequently, XTANDI® was approved by the FDA on August 31, 2012 for the treatment of patients with metastatic CRPC who had previously received TAXOTERE®. XTANDI® improved median OS and reduced the risk of death by 37% when compared to placebo. Even though these two anti-androgen therapies improved OS in metastatic CRPC patients previously treated with TAXOTERE®, the proper sequence of administration of these two agents after TAXOTERE® failure, has remained unclear. At least 2 published studies have shown that the use of ZYTIGA® as third line therapy after progression on TAXOTERE® and XTANDI® resulted in inferior outcomes.
They include ZYTIGA® (Abiraterone) and XTANDI® (Enzalutamide). ZYTIGA® inhibits CYP 17A1 enzyme thus decreasing androgen biosynthesis and depletes adrenal and intratumoral androgens. XTANDI® competes with Testosterone and Dihydrotestosterone and avidly binds to the Androgen Receptor (AR), thereby inhibiting AR signaling and in addition inhibits translocation of the AR into the nucleus and thus inhibits the transcriptional activities of the AR. There is presently very little guidance with regards to the sequencing of these two oral agents after progression on TAXOTERE®, in patients with metastatic CRPC. ZYTIGA® was approved initially by the FDA in April 2011, for use in combination with prednisone for the treatment of patients with metastatic CRPC, who had received prior chemotherapy containing TAXOTERE®. Treatment with ZYTIGA® resulted in a 35% reduction in the risk of death and a 36% increase in median Overall Survival (OS) compared with placebo. Subsequently, XTANDI® was approved by the FDA on August 31, 2012 for the treatment of patients with metastatic CRPC who had previously received TAXOTERE®. XTANDI® improved median OS and reduced the risk of death by 37% when compared to placebo. Even though these two anti-androgen therapies improved OS in metastatic CRPC patients previously treated with TAXOTERE®, the proper sequence of administration of these two agents after TAXOTERE® failure, has remained unclear. At least 2 published studies have shown that the use of ZYTIGA® as third line therapy after progression on TAXOTERE® and XTANDI® resulted in inferior outcomes.
The purpose of this study was to evaluate the role of XTANDI® as third line therapy, in patients with metastatic CRPC, following progression on TAXOTERE® and ZYTIGA®. The authors searched large, established medical databases and selected 10 publications out of 1264 articles, in which metastatic CRPC patients were treated with XTANDI® as third line therapy, following progression on TAXOTERE® and ZYTIGA®. The study included 536 patients. The primary outcomes were PSA Response Rate of more than 50%, activity of XTANDI® based on previous response to ZYTIGA® and median Progression Free Survival (PFS) or Time To Progression. The secondary outcomes included safety and the median Duration of Response. The pooled Response Rate in this patient population with XTANDI® was 22.9%. However, in patients previously sensitive to ZYTIGA®, the Response Rate with XTANDI® was higher at 35%. The median PFS was 3.1 months and the median OS was 8.3 months. This pooled analysis gives some perspective on the initial choice of anti-androgen therapy in metastatic CRPC patients who progress on TAXOTERE®, with XTANDI® benefitting the most, in patients who respond to second line treatment with ZYTIGA®. It remains to be seen if this sequencing strategy can be confirmed in prospective trials. Enzalutamide After Docetaxel and Abiraterone Acetate Treatment in Prostate Cancer: A Pooled Analysis of 10 Case Series. Petrelli F, Coinu A, Borgonovo K, et al. Clinical Genitourinary Cancer 2015;13:193-198
PRAXBIND® – An Antidote to Reverse the Anticoagulant Effects of PRADAXA®
SUMMARY: There are presently four New Oral Anticoagulants approved in the United States for the treatment of Venous ThromboEmbolism. They include PRADAXA® (Dabigatran), which is a direct thrombin inhibitor and XARELTO® (Rivaroxaban), ELIQUIS® (Apixaban), SAVAYSA® (Endoxaban), which are Factor Xa inhibitors. Compared to COUMADIN® (Warfarin), the New Oral Anticoagulants have a rapid onset of action, wider therapeutic window, shorter half-lives (7-14 hours in healthy individuals), no laboratory monitoring and fixed dosing schedule.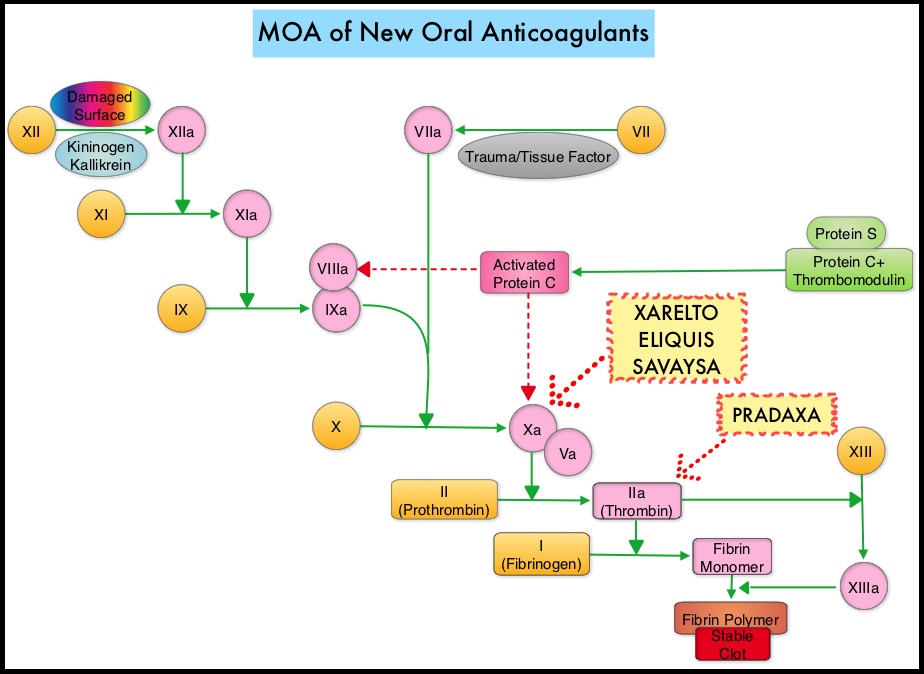 The half life of these agents can however be prolonged in those with renal insufficiency. In several clinical studies, these New Oral Anticoagulants have been shown to reduce the rate of major bleeding by 28% and the rates of intracranial and fatal hemorrhage by 50%, when compared to COUMADIN®. Unlike bleeding caused by COUMADIN®, which can be reversed using Vitamin K or Fresh Frozen Plasma, there are no specific agents presently available, for reversing bleeding caused by the New Oral Anticoagulants or for stopping the anticoagulant effects of these drugs, in patients who need urgent s
The half life of these agents can however be prolonged in those with renal insufficiency. In several clinical studies, these New Oral Anticoagulants have been shown to reduce the rate of major bleeding by 28% and the rates of intracranial and fatal hemorrhage by 50%, when compared to COUMADIN®. Unlike bleeding caused by COUMADIN®, which can be reversed using Vitamin K or Fresh Frozen Plasma, there are no specific agents presently available, for reversing bleeding caused by the New Oral Anticoagulants or for stopping the anticoagulant effects of these drugs, in patients who need urgent s
Intraperitoneal Chemotherapy Underused in Spite of Improved Survival in Advanced Ovarian Cancer
SUMMARY: The American Cancer Society estimates that over 21,000 women will be diagnosed with ovarian cancer in the United States for 2015 and over 14,000 will die of the disease. Ovarian cancer ranks fifth in cancer deaths among women, accounting for more deaths than any other cancer of the female reproductive system. Intraperitoneal (IP) delivery of antineoplatic drugs ("Belly Bath") for ovarian cancer dates back to the late 1970’s and 1980’s. This strategy for ovarian cancer was based on the fact that the peritoneal cavity is the primary site of spread and failure in most cases of advanced ovarian cancer. IP chemotherapy for ovarian cancer facilitates the exposure of tumors in the peritoneal cavity to 10-20 fold greater concentration of Cisplatin and Carboplatin and 1000 fold greater concentration of Paclitaxel, compared to IV administration, thus allowing continuous and prolonged exposure of the tumor to high drug concentrations, without systemic toxicities. Even though three Intergroup Phase III trials demonstrated the superiority of IP therapy over IV therapy, it has not been widely accepted in the US and abroad. Barriers to IP therapy have included inconvenience, IP catheter related complications, higher toxicities, lack of knowledge regarding patient selection for IP therapy as well as minimum number of cycles of IP therapy to administer and uncertain long term benefit.
The authors in this study retrospectively analyzed data from 876 patients in the two phase III, Gynecologic Oncology Group trials (GOG#114 and GOG#172). The purpose of this study was to determine the long-term survival and associated prognostic factors following IP chemotherapy, in patients with advanced ovarian cancer. In both studies, patients were randomly assigned to IP (combined N=440) or IV (combined N=436) chemotherapy. In GOG#114 trial, the two treatment groups were Paclitaxel at 135 mg/m2 IV followed by Cisplatin 75 mg/m2 IV for 6 cycles or Carboplatin IV for 2 courses followed by Paclitaxel 135 mg/m2 IV dose on day 1 and Cisplatin 100 mg/m2 IP on day 8, for 6 cycles. In GOG#172 trial, the two treatment groups (IV vs IP) were Paclitaxel at 135 mg/m2 IV followed by Cisplatin 75 mg/m2 IV on day 2 for 6 cycles or Cisplatin 100mg/m2 IP on day 2 and Paclitaxel 60 mg/m2 IP on day 8, for 6 cycles. Patients in the IP and IV groups were well balanced for baseline characteristics. At a median follow up of 10.7 years, the median Overall Survival with IP chemotherapy was 61.8 months compared with 51.4 months for IV chemotherapy and IP chemotherapy resulted in a 23% reduction in the risk of death (HR=0.77; P=0.002). IP chemotherapy was also associated with improved survival among those patients with gross residual disease ie.1 cm or less (HR = 0.75; P=0.006). The risk for death decreased by 12% for each cycle of IP chemotherapy that patients completed (HR=0.88; P<0.001). Factors significantly associated with poorer Overall Survival included clear/mucinous vs serous histology (HR=2.79; P <0 .001), gross residual vs no visible disease (HR=1.89; P< 0.001), and fewer vs more cycles of IP chemotherapy (HR=0.88; P<0.001). Younger patients were more likely to complete IP chemotherapy, with probability of completion decreasing by 5% with each additional year of age (P<0.001). The authors concluded that IP chemotherapy was associated with significantly prolonged Overall Survival in women with advanced ovarian cancer, including those with gross residual disease, when compared with IV chemotherapy. This benefit extends beyond 10 years and Overall Survival improved with increasing number of IP chemotherapy cycles administered. In a more recently published study by Wright, et al. (Wright AA, Cronin A, Milne DE, et al. Published online before print August 3, 2015, doi: 10.1200/JCO.2015.61.4776), even though the use of IP chemotherapy increased significantly at National Comprehensive Cancer Network centers between 2003 and 2012, this treatment schema was still significantly underutilized and fewer than 50% of eligible patients received it. IntraPeritoneal chemotherapy should be more often incorporated into clinical practice, to improve outcomes for patients with ovarian cancer. Long-Term Survival Advantage and Prognostic Factors Associated With Intraperitoneal Chemotherapy Treatment in Advanced Ovarian Cancer: A Gynecologic Oncology Group Study . Tewari D, Java J, Salani R, et al. JCO published online on March 23, 2015; DOI:10.1200/JCO.2014.55.9898.
FDA Approves First Biosimilar Product ZARXIO® – A Primer on Biosimilars
SUMMARY: The U.S. FDA on March 6, 2015 approved ZARXIO® (Filgrastim-sndz), the first biosimilar product approved in the United States. A biosimilar product is a biological product that is approved based on its high similarity to an already approved biological product (also known as reference product). Biological products are made from living organisms including humans, animals and microorganisms such as bacteria or yeast and are manufactured through biotechnology, derived from natural sources or produced synthetically. Biological products have larger molecules with a complex structure than conventional drugs (also known as small molecule drugs). Unlike biological products, conventional drugs are made of pure chemical substances and their structures can be identified. A generic drug is a copy of brand name drug and has the same active ingredient and is the same as brand name drug in dosage form, safety and strength, route of administration, quality, performance characteristics and intended use. Therefore, brand name and the generic drugs are bioequivalent.
The Affordable Care Act in 2010 created an abbreviated licensure pathway for biological products that are demonstrated to be “biosimilar” to, or “interchangeable” with an FDA-licensed (FDA approved) biological product (reference product). The biosimilar must show that it has no clinically meaningful differences in terms of safety and effectiveness from the reference product. A biosimilar product can only be approved by the FDA if it has the same mechanism of action, route of administration, dosage form and strength as the reference product, and only for the indications and conditions of use that have been approved for the reference product. Biosimilars are not as easy to manufacture as generics (copies of brand name drugs) because of the complexity of the structure of the biologic product and the process used to make a biologic product. The facilities where biosimilars are manufactured must also meet the FDA’s standards.
The FDA’s approval of ZARXIO® was based on review of evidence that included structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamics data, clinical immunogenicity data and other clinical safety and effectiveness data, that demonstrated ZARXIO® was biosimilar to NEUPOGEN®. ZARXIO® was approved as a biosimilar and not as an interchangeable product (Can only be substituted for the reference product after approval by the prescribing Health Care Provider). ZARXIO® is approved for the same indications as NEUPOGEN® and these indications include
• Patients with cancer receiving myelosuppressive chemotherapy
• Patients with Acute Myeloid Leukemia receiving induction or consolidation chemotherapy
• Patients with cancer undergoing Bone Marrow Transplantation
• Patients undergoing Autologous peripheral blood progenitor cell collection and therapy
• Patients with severe Chronic Neutropenia.
The most common expected side effects of ZARXIO® are bone and muscle aches, redness, swelling or itching at injection site. Less common, serious side effects include spleen rupture and serious allergic reactions. Unlike ZARXIO® which was approved via an abbreviated licensure pathway for biosimilars, GRANIX® (tbo-Filgrastim) was approved via the full Biologic License Application pathway, which presently limits GRANIX® use only for reducing the duration of severe neutropenia in patients non-myeloid malignancies, receiving myelosuppressive chemotherapy. The present Medicare reimbursement rules will be more favorable to ZARXIO® compared to GRANIX®, based on their approval process. FDA approves first biosimilar product ZARXIO®. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm436648.htm
Splanchnic Venous Thrombosis is a Marker of Cancer and a Prognostic Factor for Cancer Survival
SUMMARY: The Center for Disease Control and Prevention (CDC) estimates that approximately 1-2 per 1000 individuals develop Deep Vein Thrombosis/Pulmonary Embolism (PE) each year in the United States, resulting in 60,000 – 100,000 deaths. VTE is the third leading cause of cardiovascular mortality. Patients with unprovoked DVT and PE are two to four times more likely to be diagnosed with cancer within the following 12 months compared to the general population. It is however unknown if Splanchnic Venous Thrombosis (splanchnic veins carry blood through the liver and other abdominal organs) is a marker of occult cancer and a prognostic factor for cancer survival.
To address this question the authors from Denmark conducted a nationwide cohort study using Danish medical registries and included 1,191 patients with first time Splanchnic Venous Thrombosis (SVT) between 1994 and 2011, and followed them for subsequent cancer diagnosis, for a median of 1.6 years. They compared these results with the expected cancer risk in the general population. Additionally, to evaluate the impact of SVT on survival in those patients with cancer, the researchers compared survival in these cancer patients with a matched cohort of cancer patients without SVT. In the cohort of 1,191 patients with first time Splanchnic Venous Thrombosis (SVT), 183 patients were later diagnosed with cancer, of whom, 95 patients were diagnosed within 3 months of their SVT diagnosis. When compared to the general population, individuals diagnosed with SVT were 33 times more likely to be diagnosed with cancer within 3 months and their 3 month risk of developing cancer was 8%. The increased risk was for liver cancer, pancreatic cancer and myeloproliferative neoplasms and there was a continued twofold increase after one or more years of follow up. It was noted that Splanchnic Venous Thrombosis was also a poor prognostic factor for survival, in patients with liver and pancreatic cancer. The authors concluded that Splanchnic Venous Thrombosis (SVT) is a marker of occult malignancy, particularly liver cancer, pancreatic cancer and myeloproliferative neoplasms and SVT diagnosed in patients with liver or pancreatic cancer remains a poor prognostic factor. Therefore, patients presenting with Splanchnic Venous Thrombosis may require a more thorough diagnostic evaluation. Splanchnic venous thrombosis is a marker of cancer and a prognostic factor for cancer survival. Søgaard KK, Farkas DK, Pedersen L, et al. Blood, June 2015 DOI: 10.1182/blood-2015-03-631119
Modified GEMZAR® and ABRAXANE® Combination May Preserve Efficacy with Less Toxicity in Metastatic Pancreatic Cancer
SUMMARY: The American Cancer Society estimates that in 2015, close to 49,000 people will be diagnosed with pancreatic cancer in the United States and over 40,000 people will die of the disease. Some important risk factors for pancreatic cancer include increasing age, obesity, smoking history, genetic predisposition, exposure to certain dyes and chemicals, heavy alcohol use and pancreatitis. The best chance for long term survival is complete surgical resection, although this may not be feasible in a majority of the patients, as they present with advanced disease at the time of diagnosis. Based on the National Cancer Data Base, the 5 year observed survival rate for patients diagnosed with exocrine cancer of the pancreas is 14% for those with Stage IA disease and 1% for those with Stage IV disease. The benefit of a combination of GEMZAR® (Gemcitabine) and ABRAXANE® (nab-Paclitaxel regimen in first line treatment for metastatic pancreatic cancer was established following a open-label, randomized, phase III trial (MPACT trial) in which 861 patients with metastatic pancreatic cancer were randomized to receive either a combination of GEMZAR® and ABRAXANE® (n=431) or GEMZAR® alone (n=430). The treatment regimen consisted of GEMZAR® at 1000 mg/m2 IV and ABRAXANE® 125 mg/m2 IV, both administered on days 1, 8, and 15 of a 28 day cycle. There was a statistically significant prolongation of Overall Survival (OS) for patients in the combination group with a 28% reduction in the risk of death [HR= 0.72; P < 0.0001]. The median OS was 8.5 months in the combination group and 6.7 months in the single agent GEMZAR® group and the Progression Free Survival (PFS) in the combination group versus the single agent group was 5.5 months versus 3.7 months, respectively (HR= 0.69; P < 0.0001). In this study however, only 71% of the ABRAXANE® doses and 63% of the GEMZAR® doses were full doses, due to associated toxicities and 17% of the patients had grade 3 or 4 neuropathy.
To circumvent this toxicity, the authors at their institution adopted a modified regimen of GEMZAR® and ABRAXANE® for a similar patient population and the regimen consisted of GEMZAR® 1000 mg/m2 IV and ABRAXANE® 125 mg/m2 IV, both administered on days 1 and 15 of a 28 day cycle. They conducted a retrospective analysis of a prospectively maintained database of 69 patients treated with this modified regimen. A total of 47 patients were evaluable for responses and 63 patients were evaluable for toxicities. The median Progression Free Survival was 4.8 months and median Overall Survival was 11.1 months with the modified regimen. More importantly, the rate of grade 3 or 4 neuropathy was less than 2%. The rate of grade 3 or 4 neutropenia was 10%, and growth factor support was required in only 8% of the patients, compared with 26% for those in the MPACT trial. The authors concluded that a less intense biweekly regimen of GEMZAR® and ABRAXANE® preserves efficacy with significantly less toxicity as well as cost savings and should be a consideration, in the first line treatment of patients with metastatic pancreatic cancer. This study received a Merit Award at the 2015 Gastrointestinal Cancers Symposium. Modified gemcitabine and nab-paclitaxel in patients with metastatic pancreatic cancer (MPC): A single-institution experience. Krishna K, Blazer MA, Wei L, et al. J Clin Oncol 33, 2015 (suppl 3; abstr 366)