
The FDA on October 28, 2015 approved YERVOY® injection for the additional indication of adjuvant treatment of patients with Cutaneous Melanoma, with pathologic involvement of regional lymph nodes of more than 1 mm, who have undergone complete resection, including total lymphadenectomy. YERVOY® is a product of Bristol-Myers Squibb Company.
Month: November 2015
YONDELIS® (Trabectedin)
The FDA on October 23, 2015 approved YONDELIS®) for the treatment of patients with unresectable or metastatic Liposarcoma or Leiomyosarcoma who have received a prior Anthracycline-containing regimen. YONDELIS® injection is a product of Janssen Biotech, Inc.
ONIVYDE® (Irinotecan liposome injection)
The FDA on October 22, 2015 approved ONIVYDE® injection, administered in combination with Fluorouracil (5-FU) and Leucovorin, for the treatment of patients with metastatic Adenocarcinoma of the Pancreas, whose disease has progressed following Gemcitabine-based therapy. ONIVYDE® injection is a product of Merrimack Pharmaceuticals, Inc
PRAXBIND® (Idarucizumab)
The FDA on October 16, 2015 granted accelerated approval to PRAXBIND® for the treatment of patients treated with PRADAXA® (Dabigatran), when reversal of the anticoagulant effects of PRADAXA® is needed for emergency surgery/urgent procedures, or in life-threatening or uncontrolled bleeding. PRAXBIND® injection is a product of Boehringer Ingelheim Pharmaceuticals, Inc.
FDA Approves OPDIVO® for Renal Cell Carcinoma
SUMMARY: The FDA on November 23, 2015 approved OPDIVO® (Nivolumab) for the treatment of advanced Renal Cell Carcinoma in patients who have received prior anti-angiogenic therapy. The American Cancer Society estimates that about 61,560 new cases of kidney cancer will be diagnosed in the United States in 2015 and over 14,000 patients will die from this disease. The understanding of the Immune checkpoints has lead to the development of novel immunotherapies. Immune checkpoints or gate keepers are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. Survival of cancer cells may be related to their ability to escape immune surveillance, by inhibiting T lymphocyte activation. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies have been developed that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as PD-1(Programmed cell Death-1), etc. Following inhibition of PD-1 by specific antibodies, T cells are unleashed, resulting in T cell proliferation and activation with subsequent therapeutic responses. OPDIVO® (Nivolumab) is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the T cells. OPDIVO® in previously reported studies demonstrated an Objective Response Rate (ORR) of 20-22% and an Overall Survival (OS) of 18-25 months in previously treated patient with metastatic Renal Cell Carcinoma. AFINITOR® (Everolimus) is an inhibitor of mTOR (Mammalian Target of Rapamycin), which is a serine/threonine kinase. With the inhibition of mTOR, protein synthesis is down regulated resulting in decreased angiogenesis, cell proliferation and survival. AFINITOR® is presently indicated for the treatment of patients with advanced Renal Cell Carcinoma after failure, following treatment with SUTENT® (Sunitinib) or NEXAVAR® (Sorafenib) and has demonstrated improved median Progression Free Survival compared to placebo.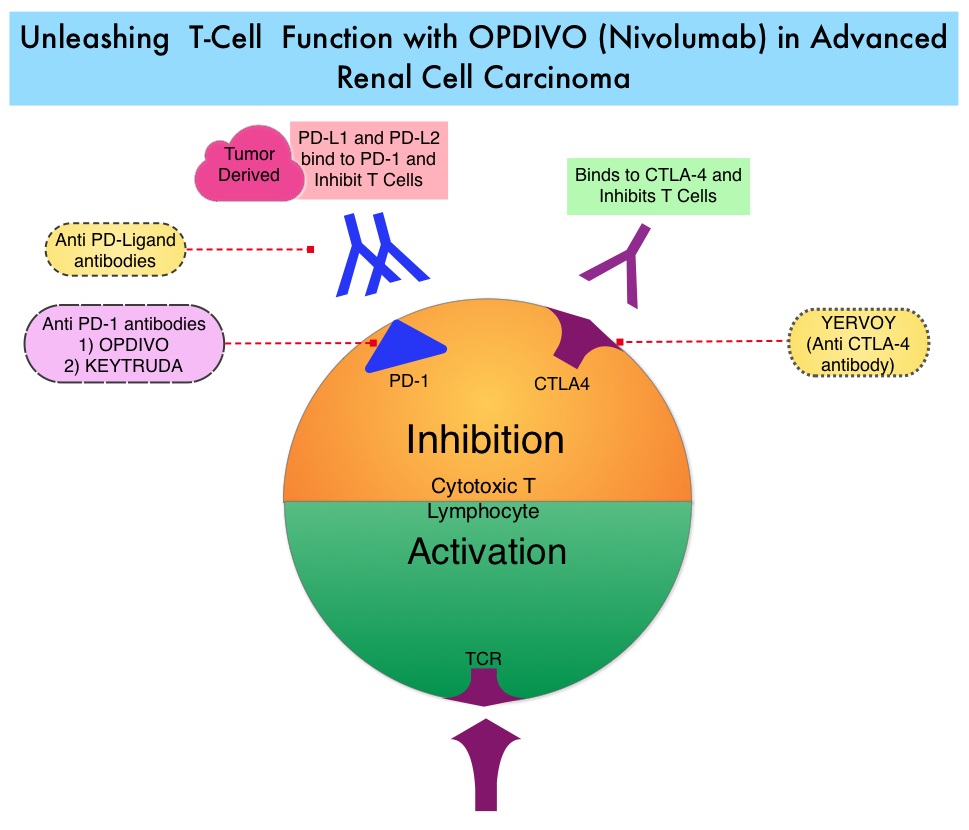
The FDA approval of OPDIVO® for Renal Cell Carcinoma (RCC), was based on a randomized, open-label, phase III study, in which 821 previously treated patients with advanced clear cell RCC, were randomly assigned in a 1:1 ratio to receive either OPDIVO® (N=410) or AFINITOR® (N=411). Treatment consisted of OPDIVO® 3 mg/kg IV every 2 weeks or AFINITOR® 10 mg PO daily. Enrolled patients had received prior treatment with one or two regimens of anti-angiogenic therapy which included SUTENT® (Sunitinib), VOTRIENT® (Pazopanib) and INLYTA® (Axitinib). The median age was 62 years. The primary end point was Overall Survival and secondary end points included Objective Response Rate and safety.
It was noted that the median Overall Survival in the OPDIVO® group was 25 months and 19.6 months in the AFINITOR® group, and this meant a 27% reduction in the risk of death with OPDIVO® (HR=0.73; P=0.002). This survival benefit was noted regardless of the PD-L1 expression level of the patient’s kidney tumors. The Objective Response Rate was greater with OPDIVO® compared with AFINITOR® (25% vs 5%; P<0.001) and the median Progression Free Survival was 4.6 months with OPDIVO® and 4.4 months with AFINITOR® (HR=0.88; P=0.11). Treatment related grade 3 or 4 adverse events occurred in 19% of the patients receiving OPDIVO® and in 37% of the patients receiving AFINITOR®.
The authors concluded that OPDIVO® significantly improved Overall Survival in patients with previously treated metastatic Renal Cell Carcinoma and is one of the few therapies with survival benefit, in this patient population. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. Motzer RJ, Escudier B, Dermott DF, et al. for the CheckMate 025 Investigators. N Engl J Med 2015; 373:1803-1813
FDA Approves First Oral Triplet Combination (NINLARO®, REVLIMID® and Dexamethasone) for Multiple Myeloma
SUMMARY: The FDA on November 20, 2015, approved NINLARO® (Ixazomib) in combination with REVLIMID® (Lenalidomide) and Dexamethasone for the treatment of patients with Multiple Myeloma who have received at least one prior therapy. Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, close to 27,000 new cases will be diagnosed in 2015 and 11,240 will die of the disease. Proteasomes are enzymes found in cells and they enable the breakdown of abnormal or mutant proteins. The amino acids from these proteins are recycled to make new proteins. Myeloma cells depend on the proteasomes to facilitate this metabolic function, to regulate their growth and survival. NINLARO® (Ixazomib) unlike VELCADE® (Bortezomib), is a second generation, oral, proteasome inhibitor, which disrupts protein metabolism in Myeloma cells, by inhibiting proteasomes and has an antiproliferative and pro-apoptotic effect.
The approval of NINLARO® was based a pivotal, multicenter, randomized, double-blind, placebo-controlled, phase III trial (TOURMALINE-MM1 study), in which 722 patients with Multiple Myeloma were randomized in a 1:1 ratio to receive either a combination of NINLARO®, REVLIMID® and Dexamethasone (N=360) or a combination of Placebo, REVLIMID® and Dexamethasone (n=362). NINLARO® was administered at 4 mg PO on days 1, 8, and 15 in combination with REVLIMID® 25 mg PO on days 1 thru 21 and Dexamethasone 40 mg PO on days 1, 8, 15, and 22 of a 28 day treatment cycle. Treatment was continued until disease progression or unacceptable toxicity. Enrolled patients had received 1 to 3 prior lines of therapy, which included VELCADE® (69%), THALOMID® (45%), and REVLIMID® (12%) and 77% of the patients had relapsed Multiple Myeloma. The median age of patients was 66 years. The primary end point of the study was Progression Free survival (PFS) and secondary endpoints included Objective Response Rate (ORR), safety, and Overall Survival.
At a prespecified interim analysis, the median PFS with the combination arm of NINLARO®, REVLIMID® and Dexamethasone was 20.6 months compared with 14.7 months for the combination group of Placebo, REVLIMID® and Dexamethasone (HR= 0.74, P=0.012).Secondary end points data was not mature at the time of this analysis. Patients in the NINLARO® group experienced more adverse events which included cytopenias, vomiting, diarrhea, peripheral neuropathy and skin rash.
The authors concluded that NINLARO® based oral triplet therapy significantly prolonged Progression Free Survival compared with REVLIMID® and Dexamethasone, with acceptable toxicities. Studies are underway, evaluating NINLARO® in newly diagnosed Myeloma patients as well as maintenance therapy in non-transplant patients. Ixazomib, an Investigational Oral Proteasome Inhibitor (PI), in Combination with Lenalidomide and Dexamethasone (IRd), Significantly Extends Progression-Free Survival (PFS) for Patients (Pts) with Relapsed and/or Refractory Multiple Myeloma (RRMM): The Phase 3 Tourmaline-MM1 Study (NCT01564537). Moreau P, Masszi T, Grzasko N, et al. 2015 ASH Annual Meeting; Orlando, FL; December 5-8, 2015. Abstract 727.
OPDIVO® (Nivolumab)
The FDA on October 9, 2015 approved OPDIVO® for the treatment of patients with metastatic Non Small Cell Lung Cancer (NSCLC), with progression on or after Platinum-based chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations, prior to receiving OPDIVO®. OPDIVO® Injection is a product of Bristol-Myers Squibb Company.
FDA Approves TAGRISSO®, a Third Generation TKI, for EGFR T790M-Positive Non Small Cell Lung Cancer
SUMMARY: The U.S. FDA granted accelerated approval to TAGRISSO® (Osimertinib), for the treatment of patients with metastatic Epidermal Growth Factor Receptor (EGFR) T790M mutation-positive Non Small Cell Lung Cancer (NSCLC), as detected by an FDA-approved test, who had progressed on or after EGFR Tyrosine Kinase Inhibitor (TKI) therapy. Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. It is the leading cause of cancer death among both men and women. The American Cancer Society estimates that over 221,200 new cases of lung cancer will be diagnosed in the United States in 2015 and over 158,000 patients will die of the disease. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer. Approximately 10% to 15% of Caucasian patients and 50% of Asian patients with Adenocarcinomas, harbor activating EGFR (Epidermal Growth Factor Receptor) mutations and 90% of these mutations are either Exon 19 deletions or L858R point mutations in Exon 21. EGFR Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib), IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), have demonstrated a 60% to 70% response rate as monotherapy when administered as first line treatment, in patients with metastatic NSCLC, who harbor the sensitizing EGFR mutations. However, majority of these patients experience disease progression within 9 to 14 months. This resistance to frontline EGFR TKI therapy has been attributed to acquired T790M “gatekeeper” point mutation in EGFR, identified in 50% – 60% of patients. The approval of TAGRISSO® was based on two multicenter, single arm, open label clinical trials (AURA and AURA2), in patients with metastatic EGFR T790M mutation-positive NSCLC, who had progressed on prior systemic therapy, including an EGFR TKI.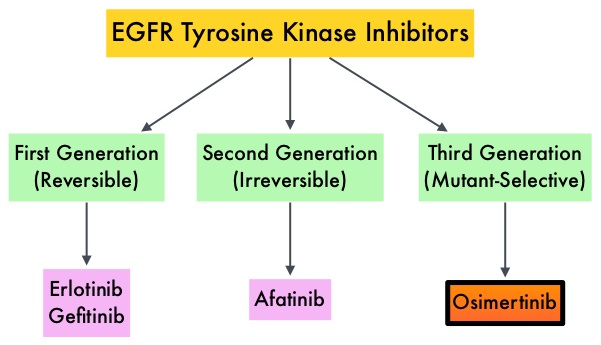
In the AURA dose escalation/expansion study (Study 1), 201 patients with EGFR mutation-positive advanced NSCLC received TAGRISSO® 80 mg PO daily until disease progression. Tumor samples were taken from all patients after disease progression on the most recent line of therapy, for prospective confirmation of T790M positive status by central laboratory testing, before enrollment. The median age was 62 years. The primary endpoint was Objective Response Rate (ORR) and secondary endpoints included Disease Control Rate (DCR), duration of response (DoR) and Progression Free Survival (PFS). The ORR in an updated analysis at the 2015 WCLC was 61% and DCR was 92%. The ORRs were similar across all lines of therapy, ie. Second line vs third line or more. The median DoR and median PFS have not been reached.
In the AURA2 Phase II study, 210 patients with locally advanced or metastatic NSCLC received TAGRISSO® 80 mg PO daily until disease progression. All eligible patients progressed on a previous EGFR TKI treatment and had a mandatory tumor sample taken after disease progression on the most recent line of therapy, for confirmation of T790M positive status by central laboratory testing. The median age was 64 years. The primary endpoint was Objective Response Rate (ORR) and secondary end points included Disease Control Rate (DCR), Duration of Response (DoR), Progression Free Survival (PFS), and safety. The ORR in an updated analysis presented at the 2015 WCLC was 71%, with 2 complete responses. The stable disease rate at 6 weeks or more was 21%, for a Disease Control Rate of 92%. The median Duration of Response was 7.8 months. The median Progression Free Survival (PFS) was 8.6 months.
Grade 1-2 toxicities from these two trials, which included a total of 411 patients included diarrhea, rash, dry skin, nail toxicity, eye disorders, nausea, decreased appetite and constipation. From these two studies it was concluded that TAGRISSO® is a new treatment option for patients who test positive for the EGFR resistance mutation, T790M, with significant response rates noted in over 50% of the treated patients. AZD9291 in pre-treated T790M positive advanced NSCLC: AURA2 Phase II study. Mitsudomi T, Tsai C, Shepherd F, et al. Presented at: 16th World Conference on Lung Cancer; September 6-9; Denver, CO. Abstract 1406.
FDA Approves YONDELIS® for Soft Tissue Sarcomas
SUMMARY: The U.S. FDA approved YONDELIS® (Trabectedin) for the treatment of patients with unresectable or metastatic Liposarcoma or Leiomyosarcoma, who have received a prior Anthracycline-containing regimen. Soft Tissue Sarcomas are a heterogeneous group of tumors of mesenchymal origin with over 50 different histological variants. The American Cancer Society estimates that in 2015, about 11,930 new soft tissue sarcomas will be diagnosed in the United States and 4,870 patients will die of the disease. The most common types of Soft Tissue Sarcomas in adults are undifferentiated pleomorphic sarcoma (previously called Malignant Fibrous Histiocytoma), Liposarcoma, and Leiomyosarcoma. Chemotherapy is the standard of care for advanced Soft Tissue Sarcomas. Following first line therapy with Anthracycline or Ifosfamide based chemotherapy regimens, Response Rates are low and second line treatment options are limited. YONDELIS® (Trabectedin) originally isolated from the Caribbean sea sponge (Ecteinascidia turbinate) is a synthetic alkaloid that binds to the minor groove of DNA and induces apoptosis by damaging the DNA.
Based on promising phase II trials, a randomized, open-label, multicenter, phase III trial was conducted, in which 518 patients with unresectable, locally advanced or metastatic Liposarcoma or Leiomyosarcomas were randomly assigned in a 2:1 to receive either YONDELIS® or Dacarbazine. YONDELIS® was dosed at 1.5 mg/m2, administered as an intravenous infusion over 24 hours (N=345) and Dacarbazine was dosed at 1000 mg/m2 administered as an intravenous infusion over 20 to 120 minutes (N=173) and the treatment was given once every 3 weeks. The median age was 56 years and enrolled patients were heavily pretreated and had received prior Anthracycline containing chemotherapy regimens. Close to 90% of the patients had received at least two prior lines of chemotherapy. Patients were stratified by Soft Tissue Sarcoma subtype (Leiomyosarcoma vs Liposarcoma), ECOG performance status, and number of prior chemotherapy regimens. The primary end point was Overall Survival (OS) and secondary end points included Progression Free Survival (PFS), Time To Progression, Objective Response Rate, Duration of Response, as well as safety.
This study demonstrated a statistically significant improvement in PFS in the YONDELIS® group with a 45% reduction in the risk of disease progression or death compared with Dacarbazine (HR= 0.55; P<0.001). The median PFS was 4.2 and 1.5 months in the YONDELIS® and Dacarbazine groups, respectively. This benefit was noted in patients with both Leiomyosarcoma and Liposarcoma. The greatest benefit in median PFS (5.6 months vs 1.5 months with YONDELIS® vs Dacarbazine, respectively) was noted in the myxoid or round cell Liposarcomas, which are considered as translocation-related sarcomas. This additional benefit can be explained based on the direct inhibition by YONDELIS®, of the chimeric FUS-CHOP translocation-generated oncoprotein, which regulates transcriptional activity in myxoid or round cell Liposarcomas. There was however no significant difference in the median Overall Survival between YONDELIS® and Dacarbazine (12.9 months vs 12.4 months, P=0.37). This may be due to crossover of close to 30% of patients who progressed on Dacarbazine to Receptor Tyrosine Kinase Inhibitor, VOTRIENT® (Pazopanib), which was approved in the U.S. during the course of this study. Most of patients who benefited from YONDELIS®, experienced stable disease as their best response for longer durations, compared to those with Dacarbazine (51% vs 35%).
The most common adverse reactions (20% or more) with YONDELIS® were nausea, fatigue, vomiting, constipation, decreased appetite, diarrhea, peripheral edema, dyspnea, and headache. The most common grade 3 to 4 adverse effects were myelosuppression and transient elevation of transaminases. The authors concluded that YONDELIS® significantly improves Progression Free Survival with superior disease control compared to Dacarbazine, in patients with heavily pretreated, advanced Liposarcoma and Leiomyosarcoma. Efficacy and Safety of Trabectedin or Dacarbazine for Metastatic Liposarcoma or Leiomyosarcoma After Failure of Conventional Chemotherapy: Results of a Phase III Randomized Multicenter Clinical Trial. Demetri GD, von Mehren M, Jones RL, et al. J Clin Oncol, doi: 10.1200/JCO.2015.62.4734.
IMLYGIC® a Novel Oncolytic Immunotherapy Demonstrates Significant Responses in Advanced Melanoma
SUMMARY: The FDA on October 27, 2015 approved IMLYGIC® (Talimogene laherparepvec or T-VEC), the first FDA-approved oncolytic virus therapy, for the treatment of melanoma lesions in the skin and lymph nodes. The American Cancer Society’s estimates that for 2015, approximately 74,000 new melanomas will be diagnosed in the United States and about 10,000 people are expected to die of the disease. IMLYGIC® is a genetically modified, herpes simplex virus type 1–derived oncolytic immunotherapy designed to induce both local and systemic immune responses. Following injection directly into melanoma lesions, IMLYGIC® selectively replicates within tumors and produces an immunostimulatory protein called Granulocyte-Macrophage Colony Stimulating Factor (GM-CSF). IMLYGIC® causes cell lysis resulting in the release of tumor-derived antigens, which along with the local GM-CSF, recruit and activate antigen-presenting cells, with subsequent induction of tumor-specific T-cell responses. The enhanced systemic antitumor immune response against tumor-derived antigens, eradicates tumor cells elsewhere in the body.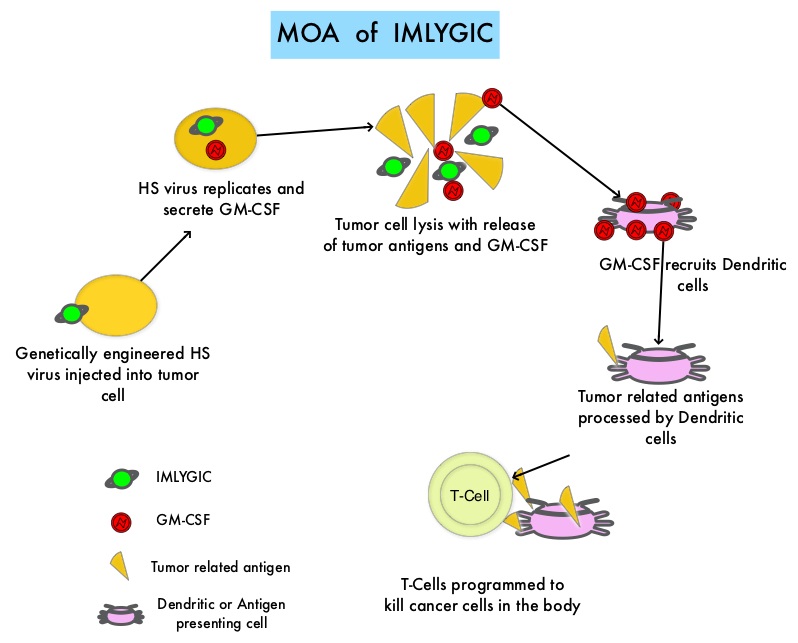
Previously reported single arm phase II study with IMLYGIC® demonstrated an Overall Response Rate (ORR) of 26% in patients with stage IIIC to IV melanoma, with responses noted in both injected and un-injected lesions, including visceral lesions. Oncovex (GM-CSF) Pivotal Trial in Melanoma (OPTiM) is a phase III study in which 436 patients with stage IIIB to IV melanoma, with injectable melanoma lesions that could not be surgically resected, were randomly assigned in a 2:1 ratio to receive intralesional IMLYGIC® (N=295) or subcutaneous GM-CSF (N=141). The enrolled patient’s melanoma lesions in the skin and lymph nodes were treated with IMLYGIC® or a comparator therapy and Injection into visceral lesions was not allowed. Patients in the IMLYGIC® group received a series of injections into the melanoma lesions. Following the initial injection, a second dose was administered three weeks later, followed by additional doses every two weeks for at least six months, unless other treatment was required or until there are were no remaining injectable lesions to treat. Patients in the GM-CSF group received 125 micrograms/m2 subcutaneously once daily for 14 days in 28-day cycles. The median age of patients in the study was 63 years. The primary end point was Durable Response Rate (DRR- Objective Response lasting continuously for 6 months or more). Secondary end points included Overall Survival (OS) and Overall Response Rate.
The DRR was significantly higher among patients receiving IMLYGIC® than among those given GM-CSF (16.3% vs 2.1%; P<0.001). The Overall Response Rate was also higher with IMLYGIC® compared to GM-CSF (26.4% vs 5.7%; P<0.001). Approximately 11% of patients receiving IMLYGIC® experienced a Complete Response compared to less than 1% for those receiving GM-CSF. The median Time to Treatment Failure was 8.2 months with IMLYGIC® and 2.9 months with GM-CSF (HR=0.42) and median Overall Survival was 23.3 months and 18.9 months, respectively (HR=0.79; P=0.051). In an exploratory subset analysis, the benefit with IMLYGIC® was more pronounced among patients with stage IIIB, IIIC, or IV M1a disease, as well as among patients who were treatment naïve. The most common side effects were fatigue, chills, fever, nausea, flu-like symptoms and pain at the injection site. IMLYGIC® should not be given to individuals with suppressed immune systems or who are pregnant because IMLYGIC® is a modified live oncolytic herpes virus therapy and herpes virus infection can potentially occur.
The authors concluded that IMLYGIC® is the first oncolytic immunotherapy to demonstrate therapeutic benefit in patients with advanced unresectable melanoma and may be another novel therapeutic option for patients with metastatic melanoma. Combining IMLYGIC® with T-Cell checkpoint inhibitor, YERVOY® (Ipilimumab) is presently being explored and thus far has shown encouraging results with minimal added toxicities. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. Andtbacka RH, Kaufman HL, Collichio F, et al. Published online before print May 26, 2015, doi: 10.1200/JCO.2014.58.3377