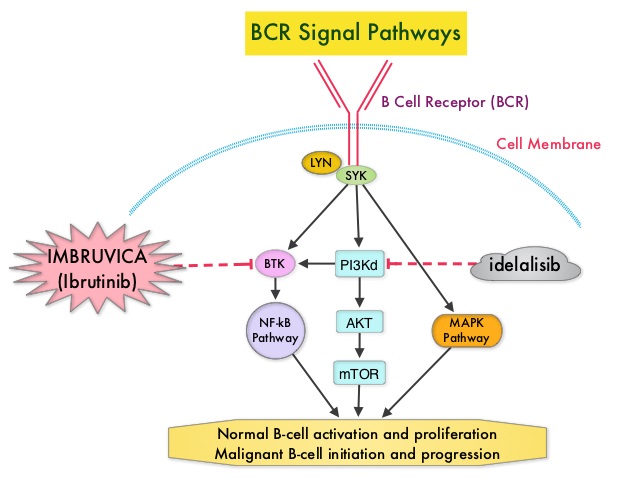
SUMMARY: The FDA on January 19, 2016 approved ARZERRA® (Ofatumumab) for extended treatment of patients who are in complete or partial response after at least two lines of therapy for recurrent or progressive Chronic Lymphocytic Leukemia (CLL). The American Cancer Society estimates that approximately 14,620 new cases of Chronic Lymphocytic Leukemia (CLL) were diagnosed in 2015 and approximately 4650 patients died from the disease. CLL is a disease of the elderly and the average age at the time of diagnosis is 72 years. ARZERRA® was previously approved for the treatment of treatment naive patients with CLL for whom FLUDARA® (Fludarabine) based therapy was considered inappropriate and also for patients with CLL refractory to FLUDARA® and CAMPATH® (Alemtuzumab). ARZERRA® is a second generation fully human IgG 1 monoclonal antibody. Unlike RITUXAN® (Rituximab), which is a chimeric monoclonal antibody, ARZERRA® targets a different region (different epitope) of the CD20 molecule. Monoclonal antibodies targeting CD20 destroy CD20 positive B cells by 3 different mechanisms. They include Antibody Dependent Cellular Cytotoxicity (ADCC), Complement Dependent Cytotoxicity (CDC) and programmed cell death (Apoptosis). Unlike RITUXAN®, ARZERRA® targets the small loop epitope of CD20 molecule which is proximal to the B cell membrane and this has been shown to be associated with highly efficient cell lysis through Complement Dependent Cytotoxicity. So, compared to RITUXAN®, ARZERRA® has stronger CDC, similar ADCC and does not appear to induce Apoptosis.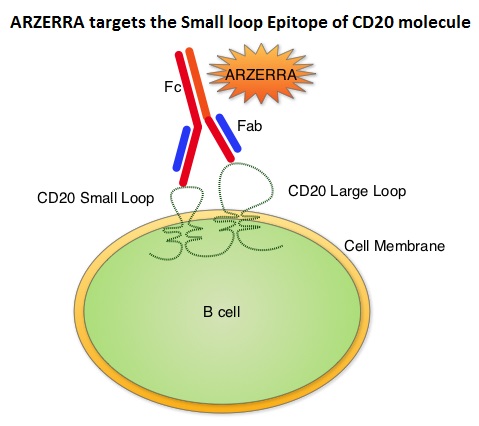
The PROLONG trial is an open-label, multicentre, randomised phase III study in which 474 patients with CLL whose disease had a complete or partial response after at least two lines of prior therapy, were randomly assigned in a 1:1 ratio to ARZERRA® (N=238) or observation (N=236). Patients in the ARZERRA® group received an initial dose of 300 mg given as an IV infusion followed by 1000 mg IV on Day 8. They subsequently received ARZERRA® 1000 mg IV every 8 weeks for up to 2 years. The median age was 65 years. The baseline characteristics in both treatment groups were well balanced. The primary endpoint was Progression Free Survival (PFS). Secondary endpoints included duration of response, Overall Survival, and safety. The median follow-up was 19•1 months. The median PFS with maintenance ARZERRA® was 29.4 months compared with 15.2 months in the observation group. This meant a 50% reduction in the risk of progression with maintenance ARZERRA® compared to observation (HR=0.50; P< 0.0001).
The most common adverse reactions in the ARZERRA® group were infusion reactions, neutropenia and upper respiratory tract infections. It was concluded that ARZERRA® is an important and new maintenance strategy in patients with relapsed CLL, to help delay disease progression. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. van Oers MHJ, Kuliczkowski K, Smolej L, et al. The Lancet Oncology 2015: 16;1282-1284
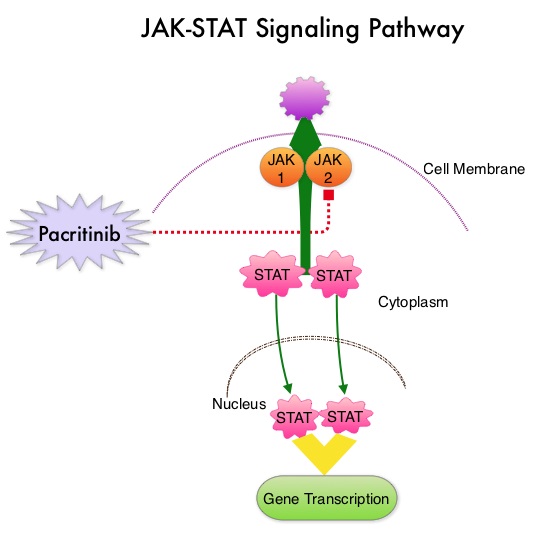 The JAK-STAT signaling pathway has been implicated in the pathogenesis of Myelofibrosis. This pathway normally is responsible for passing information from outside the cell through the cell membrane to the DNA in the nucleus, for gene transcription. Janus Kinase (JAK) family of tyrosine kinases are cytoplasmic proteins and include JAK1, JAK2, JAK3 and TYK2. JAK1 helps propagate the signaling of inflammatory cytokines whereas JAK2 is essential for growth and differentiation of hematopoietic stem cells. These tyrosine kinases mediate cell signaling by recruiting STAT’s (Signal Transducer and Activator of Transcription), with resulting modulation of gene expression. In patients with MPN, the aberrant myeloproliferation is the result of dysregulated JAK2-STAT signaling as well as excess production of inflammatory cytokines associated with this abnormal signaling. These cytokines contribute to the symptoms often reported by patients with MF. JAK2 mutations such as JAK2 V617F are seen in approximately 60% of the patients with PMF and ET and 95% of patients with PV. Unlike CML where the BCR-ABL fusion gene triggers the disease, JAK2 mutations are not initiators of the disease and are not specific for MPN. Further, several other genetic events may contribute to the abnormal JAK2-STAT signaling.
The JAK-STAT signaling pathway has been implicated in the pathogenesis of Myelofibrosis. This pathway normally is responsible for passing information from outside the cell through the cell membrane to the DNA in the nucleus, for gene transcription. Janus Kinase (JAK) family of tyrosine kinases are cytoplasmic proteins and include JAK1, JAK2, JAK3 and TYK2. JAK1 helps propagate the signaling of inflammatory cytokines whereas JAK2 is essential for growth and differentiation of hematopoietic stem cells. These tyrosine kinases mediate cell signaling by recruiting STAT’s (Signal Transducer and Activator of Transcription), with resulting modulation of gene expression. In patients with MPN, the aberrant myeloproliferation is the result of dysregulated JAK2-STAT signaling as well as excess production of inflammatory cytokines associated with this abnormal signaling. These cytokines contribute to the symptoms often reported by patients with MF. JAK2 mutations such as JAK2 V617F are seen in approximately 60% of the patients with PMF and ET and 95% of patients with PV. Unlike CML where the BCR-ABL fusion gene triggers the disease, JAK2 mutations are not initiators of the disease and are not specific for MPN. Further, several other genetic events may contribute to the abnormal JAK2-STAT signaling.
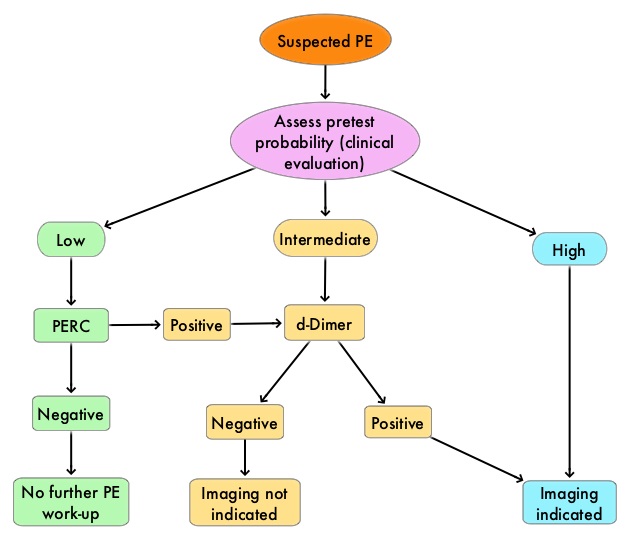
 There has also been an overuse of CT imaging and plasma d-Dimer measurement, without improvement in care, but rather harming the patient and increasing expenditure. The validated clinical decision tools, in addition to physician’s clinical judgment include the Wells and Geneva Scoring System. The PERC (see table) criteria includes 8 elements, which if absent in low risk patients rules out PE. These practice guidelines were developed to provide practical advice, based on the best available evidence.
There has also been an overuse of CT imaging and plasma d-Dimer measurement, without improvement in care, but rather harming the patient and increasing expenditure. The validated clinical decision tools, in addition to physician’s clinical judgment include the Wells and Geneva Scoring System. The PERC (see table) criteria includes 8 elements, which if absent in low risk patients rules out PE. These practice guidelines were developed to provide practical advice, based on the best available evidence.