SUMMARY: Immunotherapy in cancer management includes Cancer Vaccines, Cytokine therapy, Adoptive Cell therapy and therapy with Check Point protein inhibitors such as YERVOY®, KEYTRUDA® and OPDIVO®. Toxicities related to these immunotherapeutic interventions are mediated by T cells resulting in exaggerated T cell response and potential damage to normal tissues. A brief summary of the more common adverse events associated with cancer immunotherapy, is listed below-
CANCER VACCINES
PROVENGE® (Sipuleucel-T) is an autologous, cellular immunotherapy indicated for the treatment of asymptomatic or minimally symptomatic metastatic Castrate Resistant (hormone-refractory) Prostate Cancer. This product is the only currently approved Cancer Vaccine and consists of autologous CD54+ cells activated with recombinant PAP/GM-CSF (Prostate Acid Phosphatase, an antigen expressed in the prostate cancer tissue, linked to immune cell activator, Granulocyte Macrophage-Colony Stimulating Factor). Vaccine therapies work by promoting type 1 or type 2 immune reactions. In type 1 immune reaction, T helper type 1 (Th1) lymphocytes secrete Interleukin-2 (IL-2), Interferon gamma, and lymphotoxin-alpha and facilitate intense phagocytic activity whereas in type 2 immunity, Th2 cells secrete IL-4, IL-5, IL-9, IL-10, and IL-13 and is characterized by high antibody titers. Cancer Vaccines are associated with minimal toxicities because the antigens associated with the tumor are overexpressed in the cancer cells and are not usually detectable in normal cells. Common side effects include local reactions, fever, chills, fatigue, rash, back pain and Melanoma vaccines are associated with vitiligo.
CYTOKINE THERAPY
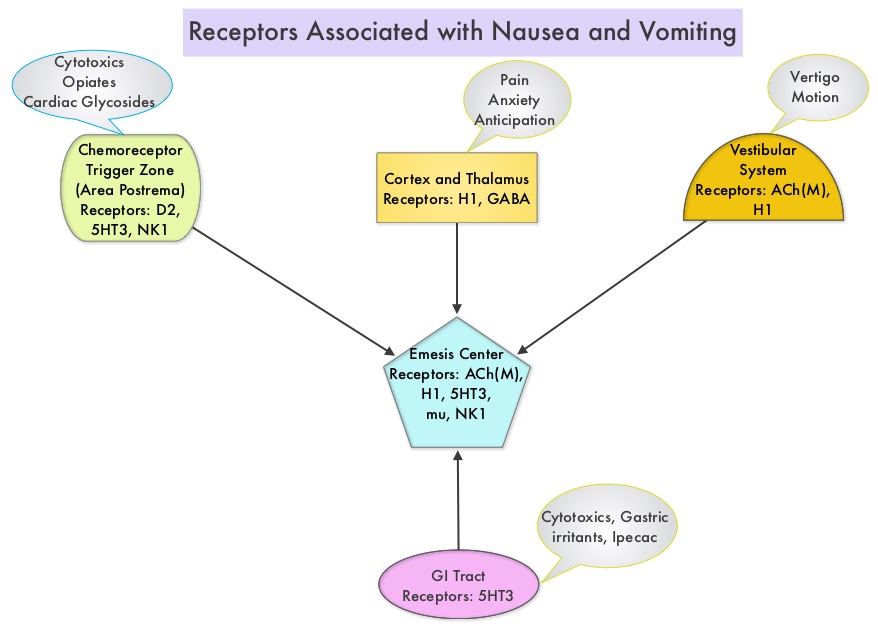
Both INTRON® A (Interferon alfa-2b) and ROFERON® A (Interferon alfa-2a) are approved for a variety of malignant conditions as well as for Chronic Hepatitis B and C. In addition to fever, chills and flu like symptoms, two thirds of the patients have nausea and anorexia and up to 45% of the patients may experience symptoms of depression. Patients should be monitored for cytopenias, diarrhea, liver toxicities as well as thyroid dysfunction and autoimmune disorders may be exacerbated with Interferon.
PROLEUKIN® (High dose IL-2) is administered in an inpatient setting with cardiac monitoring, as patients often develop capillary leak syndrome and hypotension in addition to flu like symptoms and liver function abnormalities. This has been attributed to release of Nitric Oxide, IL-1, Tumor Necrosis Factor alpha, and IFN gamma. Patients may also develop autoimmune related thyroid dysfunction, cytopenias as well as neurotoxicity and will therefore require close monitoring.
ADOPTIVE CELL THERAPY
Unlike Cancer Vaccines, Adoptive T cell therapy is a type of passive immunization which involves the transfusion of autologous or allogeneic T cells into patients with malignancies. These tumor reactive T cells can be genetically engineered or grown ex vivo and their efficacy can be enhanced by other immunotherapies, such as Cancer Vaccines, Cytokine administration or in some instances cytotoxic chemotherapy and radiation therapy. BLINCYTO® (Blinatumomab) is a genetically engineered bispecific CD19 directed CD3 T-cell engager, approved by the FDA, that binds to CD19 (expressed on B-cells) and CD3 (expressed on T-cells). It is indicated for the treatment of Philadelphia chromosome-negative relapsed or refractory B-cell precursor Acute Lymphoblastic Leukemia (ALL). Administration of BLINCYTO® or high dose IL-2 given along with T cells, can cause Cytokine Release Syndrome (CRS), associated with fever, tachycardia, vascular leak, oliguria, and hypotension. This has been attributed to IL-6 and ACTEMRA® (Tocilizumab), an IL-6 receptor antagonist may be of benefit for these patients along with IV fluids, nonsteroidal anti-inflammatory agents and vasopressors. Other toxicities that require monitoring include flu like symptoms, liver function abnormalities, B-cell aplasia, cytopenias and neurotoxicity.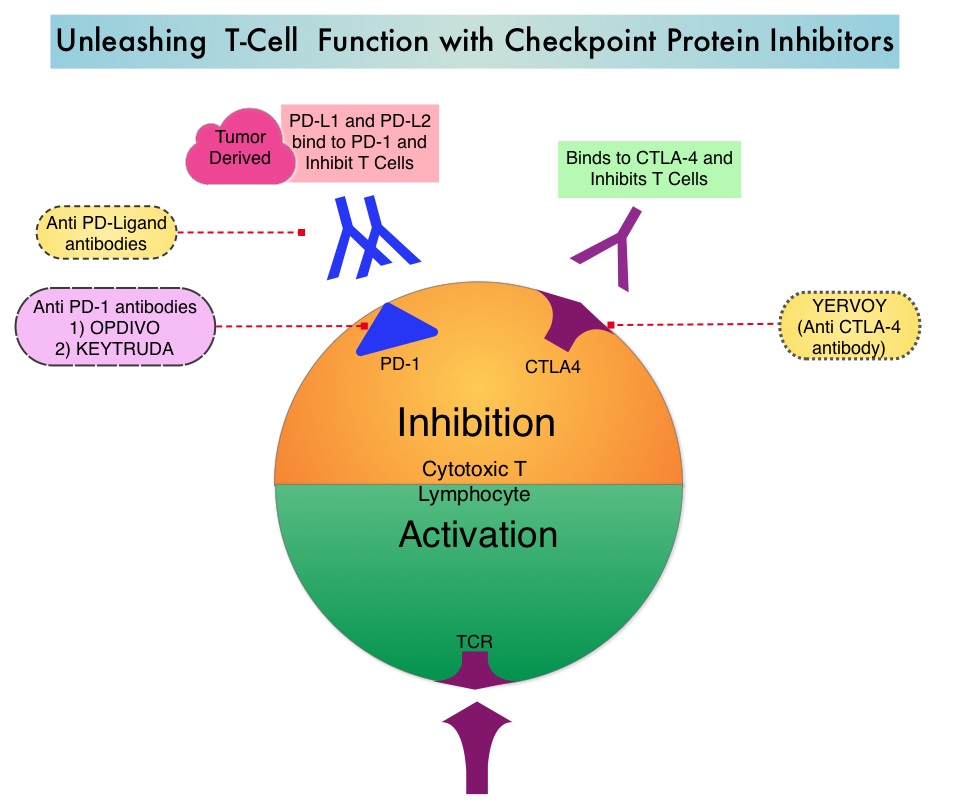
THERAPY WITH CHECKPOINT INHIBITORS
The FDA approved checkpoint inhibitors include, YERVOY® (Ipilimumab) which targets CTLA-4, KEYTRUDA® (Pembrolizumab) and OPDIVO® (Nivolumab), which block checkpoint PD-1. The toxicities associated with YERVOY® are dose dependant. Some common side effects of check point inhibitors include skin rash, flu like symptoms, liver function abnormalities, diarrhea and colitis, cytopenias, thyroid and adrenal function abnormalities. Rare cases of pneumonitis, encephalitis, Guillain-Barré syndrome, and a myasthenia gravis–like syndrome have been reported. With close monitoring, early diagnosis and intervention with Corticosteroids, these toxicities can be alleviated. REMICADE® (Infliximab), a chimeric monoclonal antibody against Tumor Necrosis Factor alpha (TNF-alpha), should be offered to those whose colitis does not resolve within 3 days of high dose steroids or for relapse of colitis with steroid taper.
Toxicities of Immunotherapy for the Practitioner. Weber JS, Yang JC, Atkins MB, et al. J Clin Oncol 2015;33:2092-2099