SUMMARY: Prostate cancer is the most common cancer in American men with the exclusion of skin cancer and 1 in 7 men will be diagnosed with prostate cancer during their lifetime. It is estimated that in the United States, about 180,890 new cases of prostate cancer will be diagnosed in 2016 and over 26,000 men will die of the disease. The development and progression of prostate cancer is driven by androgens (primarily testosterone) and androgen signaling pathways. Androgen Deprivation Therapy (ADT) has therefore been the cornerstone of treatment of advanced prostate cancer and is the first treatment intervention for hormone sensitive prostate cancer. This is accomplished by either surgical castration (bilateral orchiectomy) or medical castration using LHRH (GnRH- Gonadotropin-Releasing Hormone) agonists, given along with 2 weeks of first generation anti-androgen agents such as CASODEX® (Bicalutamide), with the anti-androgen agents given to prevent testosterone flare. There is evidence to suggest that prostate cancer cells continue to depend on androgen receptor (AR) signaling even in an androgen-deprived environment. Therefore, targeting AR and AR signaling pathways remains a rational approach in the treatment of Castration Resistant Prostate Cancer (CRPC).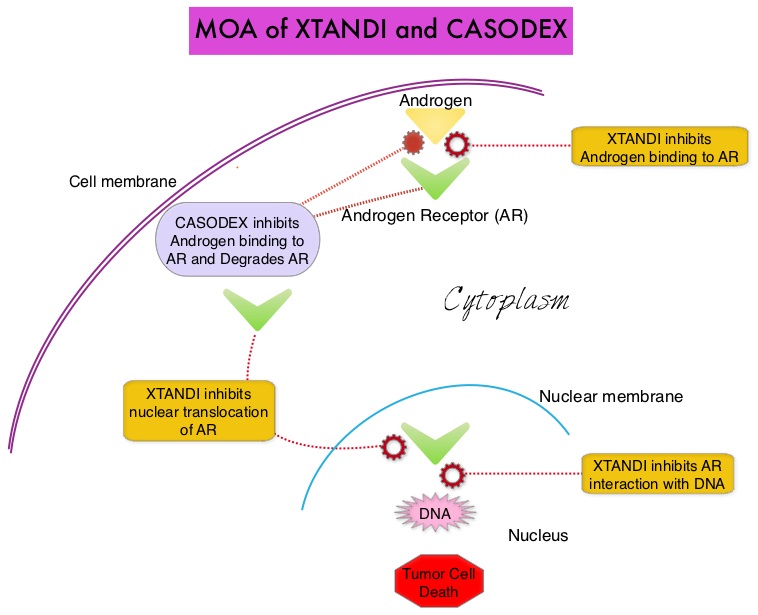
The first generation anti-androgen agents such as EULEXIN® (Flutamide), CASODEX® (Bicalutamide) and NILANDRON® (Nilutamide) act by binding to the Androgen Receptor (AR) and prevent the activation of the AR and subsequent up-regulation of androgen responsive genes. They may also accelerate the degradation of the AR. These agents have a range of pharmacologic activity from being pure anti-androgens to androgen agonists. CASODEX® is a nonsteroidal oral anti-androgen, that is often prescribed along with LHRH (GnRH- Gonadotropin-Releasing Hormone) agonists for metastatic disease or as a single agent second line hormonal therapy for those who had progressed on LHRH agonists. XTANDI® (Enzalutamide) is a second-generation anti-androgen with no reported agonistic effects. It competitively inhibits androgens and AR binding to androgens as well as AR nuclear translocation and interaction with DNA. It thus inhibits several steps in the AR signaling pathway.
TERRAIN is a double-blind, randomized phase II trial, in which 375 asymptomatic or minimally symptomatic prostate cancer patients, who had progressed following treatment with an LHRH agonists or following surgical castration, were enrolled. The objective of the TERRAIN study was to compare the efficacy and safety of XTANDI® with CASODEX®, in patients with metastatic Castration Resistant Prostate Cancer. Patients were randomly assigned in a 1:1 ratio to receive XTANDI® 160 mg daily (N=184) or CASODEX® 50 mg daily (N=191), both taken orally, in addition to Androgen Deprivation Therapy, until disease progression. Bone targeted agents, ie. Bisphosphonates and RANKL inhibitors were allowed. The primary endpoint was Progression Free Survival and secondary endpoints included PSA response and time to PSA progression. Median time on treatment for the XTANDI® group was 11.7 months and 5.8 months for the CASODEX® group.
It was noted that patients in the XTANDI® group had a significantly improved median Progression Free Survival (15.7 months) compared with 5.8 months in the CASODEX® group (HR=0.44; P<0.0001). Adverse events in the two treatment groups were different as anticipated. The most common adverse events with XTANDI® were fatigue, back pain and hot flashes whereas CASODEX® was more often associated nausea, constipation and arthralgia. Serious adverse events were experienced by 31% of the patients in the XTANDI® group and 23% of the patients in the CASODEX® group.
The authors concluded that XTANDI® increased Progression Free Survival (PFS) by nearly 10 months compared with CASODEX®, in patients with metastatic Castration Resistant Prostate Cancer (CRPC). In the PREVAIL study, XTANDI® significantly improved Overall Survival and radiographic PFS, in patients with chemotherapy-naive mCRPC and demonstrated that it can significantly delay the need for chemotherapeutic intervention. With this abundant data in favor of XTANDI®, CASODEX® may not have a significant role to play in patients with mCRPC. Efficacy and safety of enzalutamide versus bicalutamide for patients with metastatic prostate cancer (TERRAIN): a randomised, double-blind, phase 2 study. Shore ND, Chowdhury S, Villers A, et al. The Lancet Oncology 2016; 17:153-163