
SUMMARY: The FDA on May 18, 2016 approved TECENTRIQ® (Atezolizumab) for the treatment of patients with locally advanced or metastatic urothelial carcinoma, whose disease has worsened during or following platinum containing chemotherapy, or within 12 months of receiving platinum containing chemotherapy, either before (neoadjuvant) or after (adjuvant) surgical treatment. Urothelial carcinoma accounts for 90 percent of all bladder cancers and can originate in the renal pelvis, ureter and urethra. The National Cancer Institute estimates that in 2016, approximately 76,960 new cases of Bladder Cancer will be diagnosed and 16,390 patients will die of the disease. Treatment options for patients who progress after platinum based chemotherapy are limited, with poor outcomes. The treatment paradigm for solid tumors has been rapidly evolving, with a better understanding of the Immune checkpoints or gate keepers. Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. Survival of cancer cells in the human body may be to a significant extent related to their ability to escape immune surveillance, by inhibiting T lymphocyte activation. The T cells of the immune system therefore play a very important role in modulating the immune system. Under normal circumstances, Immune checkpoints or gate keepers inhibit an intense immune response by switching off the T cells of the immune system. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies are being developed that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152), PD-1(Programmed cell Death 1), as well as Programmed cell Death Ligands (PD-L1) that are expressed by cells in the tumor micro environment. By doing so, T cells are unleashed, resulting in T cell proliferation, activation and a therapeutic response.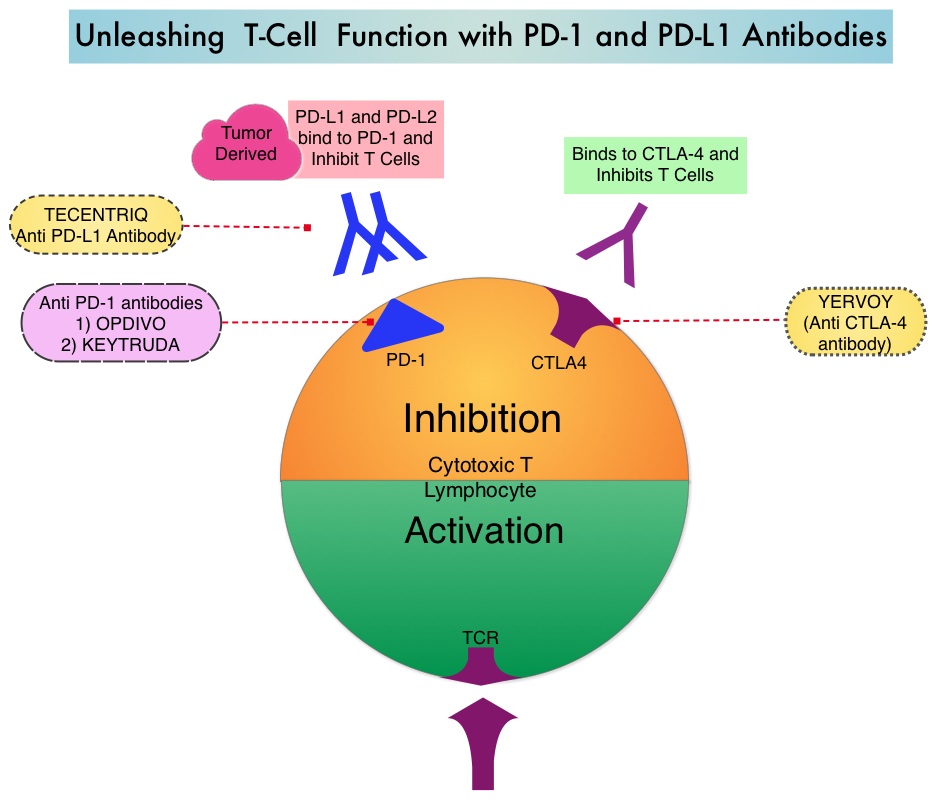
TECENTRIQ® (Atezolizumab) is an anti-PDL1 monoclonal antibody designed to directly bind to PD-L1 expressed on tumor cells and tumor-infiltrating Immune Cells, thereby blocking its interactions with PD-1 and B7.1 receptors and thus enabling the activation of T cells. The FDA approval was based on IMvigor 210 trial which is an open label, single arm, multicenter, phase II study in which the safety and efficacy of TECENTRIQ® was evaluated in patients with locally advanced or metastatic urothelial carcinoma, regardless of PD-L1 expression. In this study, 310 patients whose disease had progressed during or following previous treatment with a platinum based chemotherapy regimen received TECENTRIQ® 1200 mg as an intravenous dose once every 21 days cycles until disease progression. The primary endpoint of the study was Objective Response Rate (ORR) and secondary endpoints included Duration of Response (DOR), Overall Survival (OS), Progression Free Survival (PFS) and safety. PD-L1 expression on tumor-infiltrating Immune Cells (IC) was assessed prospectively by ImmunoHistoChemistry using Formalin-Fixed Paraffin-Embedded tumor specimens. PD-L1 positivity was defined as follows – IC 2/3 (5% or more), IC 1 (1-5%) and IC 0 (<1%).
In an updated analysis, with a median follow of 11.7 months, the ORR was 15% for the entire cohort of patients (P=0.0058). The ORR in the IC 2/3 group was 26% (P<0.0001) and 18% in the IC 1 group (P=0.0004). Ongoing responses were noted in 84% of the responding patients. The most common side effects associated with TECENTRIQ® included fatigue, decreased appetite, nausea, urinary tract infection, fever and constipation.
The authors concluded that TECENTRIQ® significantly improves Objective Response rate (ORR) in metastatic urothelial cancers and this benefit is even more so in those tumors with a higher level of PD-L1 expression. Rosenberg JE, Hoffman-Censits, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 2016; Published online 4 March. DOI: http://dx.doi.org/10.1016/S0140-6736(16)00561-4
Month: May 2016
FDA Approves OPDIVO® for Relapsed Classical Hodgkin Lymphoma
SUMMARY: The FDA on May 17, 2016, granted accelerated approval to OPDIVO® (Nivolumab) for the treatment of patients with Classical Hodgkin Lymphoma (cHL) that has relapsed or progressed after autologous Hematopoietic Stem Cell Transplantation (HSCT) and post-transplantation ADCETRIS® (Brentuximab vedotin). The American Cancer Society estimates that in the United States for 2016, about 8500 new cases of Hodgkin lymphoma will be diagnosed and over 1100 patients will die of the disease. Hodgkin lymphoma is classified into two main groups-Classical Hodgkin lymphomas and Nodular Lymphocyte Predominant type, by the World Health Organization. The Classical Hodgkin lymphomas include Nodular sclerosing, Mixed cellularity, Lymphocyte rich, Lymphocyte depleted subtypes and accounts for approximately 10% of all malignant lymphomas. Nodular sclerosis Hodgkin lymphoma histology, accounts for approximately 80% of Hodgkin lymphoma cases in older children and adolescents in the United States. Classical Hodgkin Lymphoma is a malignancy of primarily B lymphocytes and is characterized by the presence of large mononucleated Hodgkin and giant multinucleated Reed-Sternberg (RS) cells collectively known as Hodgkin and Reed-Sternberg cells (HRS). The HRS cells in turn recruit an abundance of ineffective inflammatory cells and infiltrates of immune cells. Preclinical studies suggest that HRS cells evade immune detection by exploiting the pathways associated with immune checkpoint, Programmed Death-1 (PD-1) and its ligands PD-L. Classical Hodgkin Lymphoma is an excellent example of how the tumor microenvironment influences cancer cells to proliferate and survive.
The most common genetic abnormality in Nodular sclerosis subtype of Hodgkin lymphoma is the selective amplification of genes on the short arm of chromosome 9 (9p24.1) which includes JAK-2 with resulting increased expression of PD-1 ligands such as PDL1 and PDL2 on HRS cells, as well as increased JAK-STAT activity, essential for the proliferation and survival of Hodgkin Reed-Sternberg (HRS) cells. Infection with Epstein–Barr virus (EBV) similarly can increase the expression of PDL1 and PDL2 in EBV-positive Hodgkin's lymphomas. It would therefore seem logical to block or inhibit immune check point PD-1 rather than both its ligands, PDL1 and PDL2.
Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. Under normal circumstances, Immune checkpoints or gate keepers inhibit intense immune responses by switching off the T cells of the immune system. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies are being developed that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152), PD-1(Programmed cell Death 1), etc. By doing so, one would expect to unleash the T cells, resulting in T cell proliferation, activation and a therapeutic response. OPDIVO® is an immune checkpoint PD-1 (Programmed cell Death 1) targeted, fully human, immunoglobulin G4 monoclonal antibody that demonstrated significant responses in a phase 1b trial involving patients with relapsed/refractory cHL (Ansell SM et al. NEJM 2015;372:311–319).
This accelerated approval of OPDIVO® was based on two single-arm, open label, multicenter trials (CheckMate-205 and -039) of OPDIVO®, in adults with relapsed or refractory cHL. These trials enrolled patients regardless of PD-L1 expression status on Reed-Sternberg cells. The median age was 37 years and patients had a median of 5 prior therapies. The primary endpoint was Objective Response Rate (ORR) and additional outcome measures included Duration of Response (DOR). Patients received OPDIVO® (Nivolumab) at a dose of 3 mg/kg every 2 weeks and treatment was continued until patients had a complete response, tumor progression or severe toxicities.
The efficacy of OPDIVO® was evaluated in 95 patients with cHL from the two single-arm trials, previously treated with autologous Hematopoietic Stem Cell Transplantation (aHSCT) and post-transplantation ADCETRIS® (Brentuximab vedotin). Patients had received a median of 17 doses of OPDIVO®. The ORR with OPDIVO® was 65% with 58% Partial Remission and 7% Complete Remission. The median time to response was 2.1 months and the estimated median DOR was 8.7 months. The most common adverse reactions (20% or more) of any grade were fatigue, upper respiratory tract infection, cough, pyrexia, and diarrhea.
The authors concluded that patients with Classical Hodgkin Lymphoma treated with OPDIVO® after autologous Hematopoietic Stem Cell Transplantation (aHSCT) and post-transplantation ADCETRIS®, have very high response rates and long Duration of Responses with acceptable toxicities. OPDIVO® is the first PD-1 inhibitor, approved for a hematologic malignancy. Checkmate 205: Nivolumab (nivo) in classical Hodgkin lymphoma (cHL) after autologous stem cell transplant (ASCT) and brentuximab vedotin (BV)—A phase 2 study. Younes A, Santoro A, Zinzani PL, et al. J Clin Oncol 34, 2016 (suppl; abstr 7535)
FDA Approves Combination of LENVIMA® and AFINITOR® for Advanced Renal Cell Carcinoma
SUMMARY: The FDA on May 13, 2016 approved LENVIMA® (Lenvatinib) in combination with AFINITOR® (Everolimus), for the treatment of advanced Renal Cell Carcinoma, following one prior anti-angiogenic therapy. LENVIMA® was first approved in 2015 for the treatment of locally recurrent or metastatic, progressive, radioactive iodine-refractory differentiated thyroid cancer. The American Cancer Society estimates that about 62,700 new cases of kidney cancer will be diagnosed in the United States in 2016 and over 14,000 patients will die from this disease.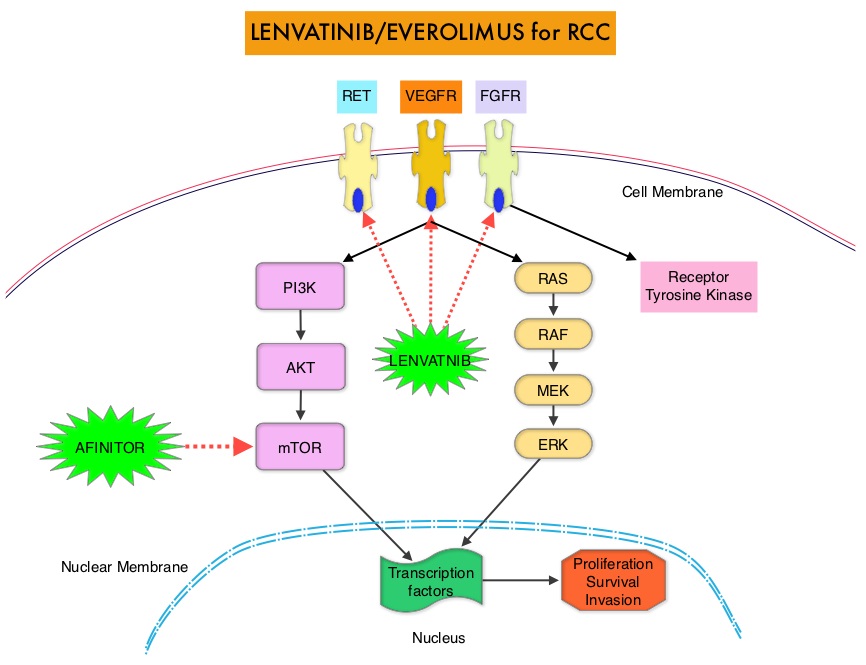
LENVIMA® is an oral multitargeted TKI (Tyrosine Kinase Inhibitor), which targets Vascular Endothelial Growth Factor Receptor (VEGFR)1-3, Fibroblast Growth Factor Receptor (FGFR)1-4, REarranged during Transfection tyrosine kinase receptor (RET), c-KIT, and Platelet Derived Growth Factor Receptor (PDGFR). LENVIMA® differs from other TKIs with anti-angiogenesis properties by its ability to inhibit FGFR-1, thereby blocking the mechanisms of resistance to VEGF/VEGFR inhibitors. In addition, it controls tumor cell growth by inhibiting RET, c-KIT, and PDGFR beta and influences tumor microenvironment by inhibiting by FGFR and PDGFR beta. AFINITOR® (Everolimus) does not inhibit tyrosine kinases, but is a specific inhibitor of mTOR (Mammalian Target of Rapamycin), which is a serine/threonine kinase, normally activated further downstream in the signaling cascade. With the inhibition of mTOR, protein synthesis is inhibited resulting in decreased angiogenesis, cell proliferation and survival as well as decreased levels of HIF-1 alpha.
Metastatic Renal Cell Carcinoma is currently treated with sequential therapy using single agent VEGF or mTOR inhibitors. The current FDA approval was based on a multicenter open-label phase II study in which 153 patients were randomized in a 1:1:1 ratio to receive LENVIMA® plus AFINITOR® (N=51), LENVIMA® monotherapy (N=52), or AFINITOR® monotherapy (N=50). Enrolled patients had advanced or metastatic Renal Cell Carcinoma and had previously received anti-angiogenic therapy. Patients in the combination group received LENVIMA® 18 mg QD along with AFINITOR® 5 mg QD, orally, whereas patients in the single agent groups received either LENVIMA® 24 mg QD or AFINITOR® 10 mg QD. Patients were not allowed to crossover in this study. Using the MSKCC (Memorial Sloan Kettering Cancer Center) risk classification, patients were well balanced between the treatment groups. The primary endpoint was Progression Free Survival. Secondary endpoints included Objective Response Rate (ORR) and Overall Survival (OS).
It was noted that the median PFS in the combination group was 14.6 months compared with 5.5 months in the AFINITOR® alone group (HR=0.37; P=0.0005). This meant a 63% reduction in the risk of disease progression. The median OS with the combination treatment was 25.5 months compared to 15.4 months with AFINTOR® alone (HR=0.67) and this meant a 33% reduction in the risk of death. The ORR with the LENVIMA® and AFINITOR® combination was 37% compared with 6% in the AFINITOR® alone group. The PFS at one year was 31% with the combination treatment versus 14% with single agent AFINITOR® and at 2 years 51% of patients remained alive in the combination treatment group compared with 26% in the single-agent AFINITOR® group. The most common grade 3-4 toxicities in the combination group included diarrhea, fatigue, arthralgia, hypertension and anemia.
The authors concluded that a combination of LENVIMA® and AFINITOR® significantly improves Progression Free Survival, in patients with metastatic Renal Cell Carcinoma, who had progressed on previous VEGF targeted therapy and this combination therapy may change the treatment paradigm for this patient population. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Motzer RJ, Hutson TE, Glen H, et al. Lancet Oncol. 2015;16:1473-1482.
FDA Approves IMBRUVICA® as Initial Therapy for Patients with Chronic Lymphocytic Leukemia
SUMMARY: The FDA on March 4, 2016 approved the expanded use of IMBRUVICA® (Ibrutinib) as first line treatment for patients with Chronic Lymphocytic Leukemia (CLL) and Small Lymphocytic Lymphoma. IMBRUVICA® received approval in February 2014 for the treatment of Chronic Lymphocytic Leukemia (CLL) in patients who received at least one prior therapy and in July 2014 for the treatment of CLL with 17p deletion. The American Cancer Society estimates that approximately 18,960 new cases of Chronic Lymphocytic Leukemia (CLL) will be diagnosed in 2016 and approximately 4660 patients will die from the disease. CLL is a disease of the elderly and the average age at the time of diagnosis is 72 years. There are two main types of lymphocytes, B and T lymphocytes/cells. B-cell CLL is the most common type of leukemia in adults. Normal B-cell activation and proliferation is dependent on B-cell receptor (BCR) signaling. This signaling is also important for initiation and progression of B-cell lymphoproliferative disorders. Bruton's Tyrosine Kinase (BTK) is a member of the Tec family of kinases, downstream of the B-cell receptor and is predominantly expressed in B-cells. It is a mediator of B-cell receptor signaling in normal and transformed B-cells. Following binding of antigen to the B-Cell Receptor, kinases such as Syk (Spleen Tyrosine Kinase), Lyn (member of the Src family of protein tyrosine kinases) and BTK (Bruton's Tyrosine Kinase) are activated, with subsequent propagation through PI3K/Akt, MAPK, and NF-κB pathways. This results in B-cell activation and proliferation. IMBRUVICA® (Ibrutinib) is an oral, irreversible inhibitor of BTK and inhibits cell proliferation and promotes programmed cell death (Apoptosis) by blocking B-cell activation and signaling. In elderly CLL patients with comorbid conditions, Chlorambucil is often considered as a standard first-line therapy because of the higher rate of toxicities associated with FLUDARA® (Fludarabine) and TREANDA® (Bendamustine).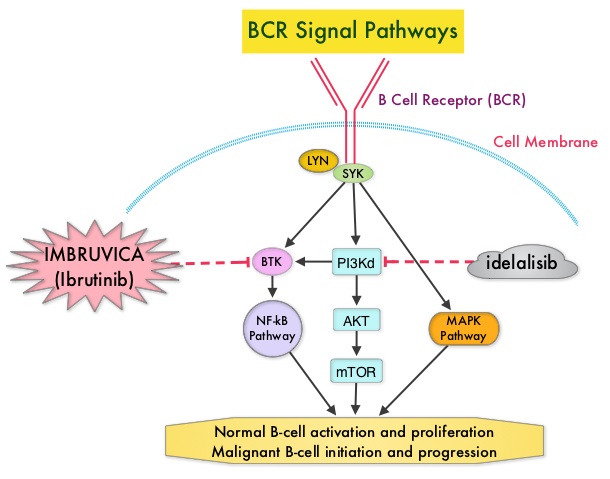
RESONATE-2 is a international, open-label, randomized, phase III trial, in which the efficacy of two oral agents , IMBRUVICA® and Chlorambucil, were compared, in previously untreated elderly patients with CLL or Small Lymphocytic Lymphoma. In this study, 269 treatment naïve patients with CLL or Small Lymphocytic Lymphoma, who were 65 years of age or older, were randomly assigned in a 1:1 ratio, to receive IMBRUVICA® 420 mg PO once daily (N=136) or Chlorambucil at a dose of 0.5 mg/kg on days 1 and 15 of each 28 day cycle, increased to a maximum of 0.8 mg/kg, if tolerated (N=133). Patients with chromosome del (17p) were excluded. The median age was 73 years and 70% of patients were over age 70. The primary end point was Progression Free Survival (PFS) and secondary end points included, Overall Response Rate (ORR), Overall Survival (OS), and sustained hematologic improvement.
With a median follow-up of 28.1 months, patients in the IMBRUVICA® group had a significantly longer Progression Free Survival (PFS) compared to the Chlorambucil group (median not reached versus 18.9 months), with a risk of progression or death 84% lower with IMBRUVICA®, compared to Chlorambucil (HR=0.16; P<0.001). IMBRUVICA® significantly prolonged Overall Survival, in spite of 41% of the patients crossing over from the Chlorambucil group to IMBRUVICA®. The 2 year Overall Survival rate was 94.7% in the IMBRUVICA® group and 84.3% in the Chlorambucil group (HR=0.44). Further, IMBRUVICA® significantly improved Overall Response Rate, compared with Chlorambucil (82% vs 35%; P< 0.0001) and also significantly improved hemoglobin and platelets levels from baseline values, compared with Chlorambucil. The most common adverse events associated with IMBRUVICA® were diarrhea, fatigue, cough and nausea and were mostly Grade 1 toxicities.
The authors concluded that IMBRUVICA® significantly improves Progression Free Survival, Overall Survival and Overall Response Rate, compared to Chlorambucil, in previously untreated patients with CLL or Small Lymphocytic Lymphoma, and should therefore be considered for all elderly patients who are not candidates for aggressive systemic therapy. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. Burger JA, Tedeschi A, Barr PM, et al. N Engl J Med 2015; 373:2425-2437
FDA Approves GILOTRIF® for Squamous Cell Carcinoma of the Lung
SUMMARY: The FDA on April 15, 2016 approved GILOTRIF® (Afatinib) tablets for the treatment of patients with advanced Squamous Cell Carcinoma of the lung, whose disease has progressed after treatment with Platinum-based chemotherapy. Lung cancer is the second most common cancer in both men and women and the American Cancer Society estimates that for 2016, about 224,390 new cases of lung cancer will be diagnosed and over 158,000 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 25% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas. Non Small Cell Lung Cancer patients with Squamous Cell histology have been a traditionally hard- to-treat, patient group, with less than 5% of patients with advanced SCC, surviving for five years or longer. Some of the advanced NSCLC tumors are dependent on the Epidermal Growth Factor Receptor (EGFR) for cell proliferation and survival, regardless of EGFR mutation status. TARCEVA® (Erlotinib) is a reversible EGFR Tyrosine Kinase Inhibitor and is presently approved by the FDA for the treatment of locally advanced or metastatic NSCLC, after failure of at least one prior chemotherapy regimen. GILOTRIF® (Afatinib) is an oral, irreversible blocker of the ErbB family which includes EGFR (ErbB1), HER2 (ErbB2), ErbB3 and ErbB4. GILOTRIF® was approved by the FDA in July 2013, for the first line treatment of patients with metastatic NSCLC, whose tumors have Epidermal Growth Factor Receptor (EGFR) exon 19 deletions or exon 21 (L858R) substitution mutations.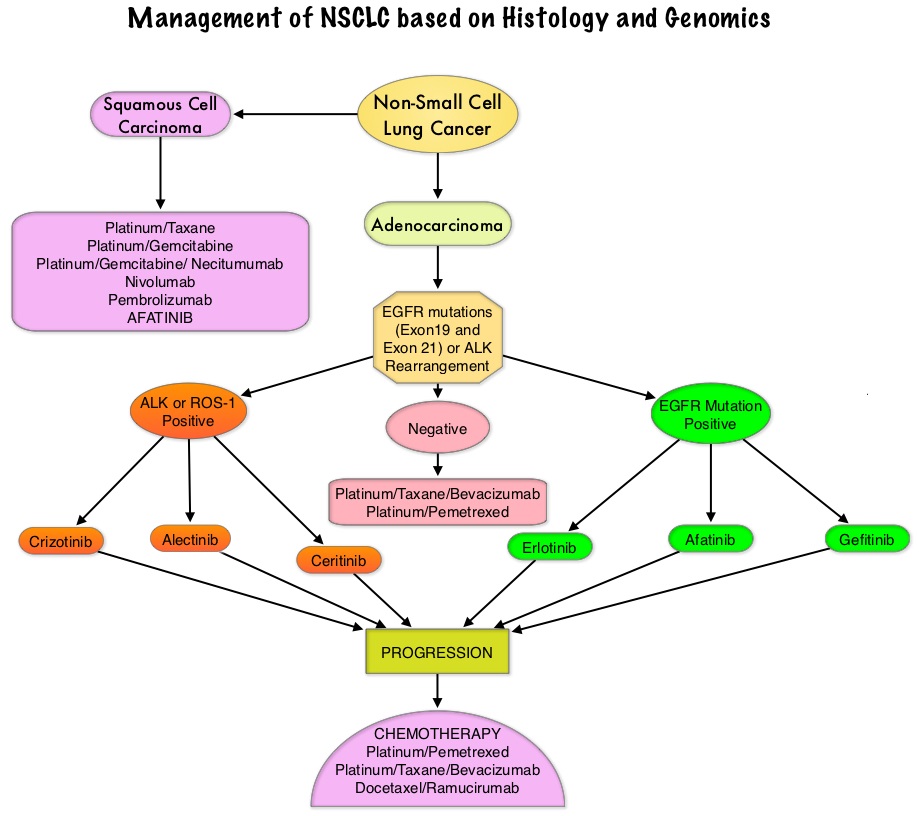
This additional indication approved by the FDA was based on the LUX-Lung 8 study, which is a phase III trial in which 795 patients with Stage IIIB/IV Squamous Cell Carcinoma of the lung who had progressed on first line platinum based doublet therapy, were randomized 1:1 to receive GILOTRIF® 40 mg PO daily (N=398) or TARCEVA® 150 mg PO daily (N=397). Treatment was given until disease progression. The median age was 65 years. Majority of the patients were male, caucasian and ex-smokers. The Primary endpoint was Progression Free Survival (PFS) and Secondary endpoints included Overall Survival (OS), Objective Response Rate (ORR), Disease Control Rate (DCR), patient reported outcomes and safety. The Primary endpoint of Progression Free Survival (PFS) was met and reported in 2014 and favored GILOTRIF® over TARCEVA®. The authors in this analysis reported the Overall Survival data, as well as updated data on Progression Free Survival and other Secondary endpoints. The median Overall Survival was 7.9 months with GILOTRIF® and 6.8 months with TARCEVA® (HR=0.81; P=0.007). This meant a 19% reduction in the risk of death with GILOTRIF® when compared to TARCEVA® and this survival advantage was consistent across all time points. The updated median Progression Free Survival for GILOTRIF® was 2.4 months versus 1.9 months for TARCEVA® (HR=0.82; P=0.04) which meant an 18% reduction in the disease progression. The Disease Control Rate was 51% for GILOTRIF® and 40% with TARCEVA® (P=0.002). Based on patient reported outcomes, symptoms including cough and dyspnea were better with GILOTRIF® compared to TARCEVA®. Incidence of severe adverse events was similar with both therapies, with patients on GILOTRIF® experiencing more grade 3 diarrhea and stomatitis and patients receiving TARCEVA® experiencing more grade 3 rash.
The authors concluded that GILOTRIF® should be the TKI of choice for the second line treatment of patients with Squamous Cell Carcinoma of the lung, as it significantly improves Overall Survival, Progression Free Survival, Disease Control Rate and controls symptoms with manageable toxicities, when compared to TARCEVA®. Afatinib versus erlotinib as second-line treatment of patients with advanced squamous cell carcinoma of the lung (LUX-Lung 8): an open-label randomised controlled phase 3 trial. Soria J, Felip E, Cobo M, et al. The Lancet Oncology 2015;16:897-907
Five Year Follow up Data without Adjuvant Chemotherapy, Utilizing Oncotype DX 21-gene Recurrence Score Assay
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately, 246,660 new cases of invasive breast cancer will be diagnosed in 2016 and 40,450 women will die of the disease. Patients with early stage breast cancer often receive adjuvant chemotherapy. The Oncotype DX breast cancer assay, is a multigene genomic test that analyzes the activity of a group of 21 genes and is able to predict the risk of breast cancer recurrence and likelihood of benefit from systemic chemotherapy, following surgery, in women with early stage breast cancer. Chemotherapy recommendations for early stage, hormone receptor positive, HER negative breast cancer patients, are often made based on tumor size, grade, immunohistochemical markers such as Ki-67, nodal status and 21-gene Recurrence Score (RS) assay.
The West German Study Group (WSG) PlanB phase III trial, used Recurrence Score assay prospectively, to define a low risk subset of patients with node negative disease with high risk traditional parameters and patients with node positive disease (HR+, HER2 negative), who could be treated with adjuvant endocrine therapy alone, sparing chemotherapy. In this study, patients with a Recurrence Score of 11 or less were defined as having low risk for recurrence, even in those considered to have tumor with high risk features such as large tumor size, high tumor grade and lymph node involvement. Patients were considered to be at intermediate or high risk if they had a Recurrence Score of 12 or more and 25 or more, respectively. Gluz and colleagues had previously reported 3-year follow up results of a planned interim analysis of this study, and were able to show significant difference between tumor grade, Ki-67 and Oncotype DX Recurrence Score.
In this current analysis, the authors reported the 5-year Disease Free Survival (DFS) outcomes of this large prospective trial. This analysis included data from 3,198 patients with early stage hormone receptor positive or HER2 negative breast cancer. The median age was 56 years and 32.5% of the patients had grade 3 tumors and 41% of the patients had node positive disease. Patients with a Recurrence Score of 11 or less received hormonal therapy and adjuvant chemotherapy was omitted. The intermediate and high risk patients were randomized to receive six cycles of TAXOTERE® (Docetaxel)/CYTOXAN® (Cyclophosphamide) or four cycles of ELLENCE® (Epirubicin)/CYTOXAN® followed by four cycles of TAXOTERE®. The primary endpoint was Disease Free Survival (DFS) defined as invasive or noninvasive relapse.
It was noted that the 5-year DFS in the low risk group was 94%, 84% in the high risk group and 94% in the intermediate risk group (P<0.001). It should be noted that approximately 15% of the patients in the clinically determined intermediate or high risk group, with 0-3 lymph node involvement, fell in the low genomic risk group (Recurrence Score of 11 or less) and received hormonal therapy alone.
The authors concluded that West German Study Group (WSG) PlanB study is the first trial that has reported five year survival data using 21-gene Recurrence Score assay, which can identify patients with early breast cancer, who would benefit from hormonal therapy alone and could be spared chemotherapy. Based on these data, 21-gene Recurrence Score has stronger prognostic value compared to immunohistochemical studies such as Ki-67 and hormone-receptor expression and should therefore be routinely incorporated in clinical practice as a decision making tool for this patient population, in addition tumor size, grade and nodal status. Prospective WSG Phase III PlanB trial: Clinical outcome at 5 year follow up and impact of 21 Gene Recurrence Score result, central/local-pathological review of grade, ER, PR and Ki67 in HR+/HER2- high risk node-negative and –positive breast cancer. Gluz O, Nitz U, Christgen M, et al. Abstract 8LBA. Presented at: 10th European Breast Cancer Conference; March 9-11, 2016; Amsterdam.
OPDIVO® Granted Breakthrough Designation by FDA for Head and Neck cancer
SUMMARY: The FDA granted breakthrough therapy designation to Nivolumab (OPDIVO®), as a treatment for patients with recurrent or metastatic Squamous Cell Carcinoma of the Head and Neck (SCCHN), following a platinum based therapy. This designation was based on the findings from CheckMate-141 study, which demonstrated an improvement in Overall Survival (OS) with OPDIVO®, compared to investigator’s choice of therapy. The American Cancer Society estimates that 61,760 people will be diagnosed with Head and Neck cancer in 2016 and 13,190 patients will die of the disease. Patients with recurrent/metastatic Squamous Cell Carcinoma of the Head and Neck (SCCHN) have a poor prognosis with a median Overall Survival (OS) of about 13 months with first line therapy and about 6 months or less with later lines of therapy. The treatment paradigm for solid tumors has been rapidly evolving with a better understanding of the Immune checkpoints or gate keepers. Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. Survival of cancer cells in the human body may be to a significant extent related to their ability to escape immune surveillance by inhibiting T lymphocyte activation. The T cells of the immune system therefore play a very important role in modulating the immune system. Under normal circumstances, inhibition of an intense immune response and switching off the T cells of the immune system, is an evolutionary mechanism and is accomplished by Immune checkpoints or gate keepers. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies are being developed that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152), PD-1(Programmed cell Death 1), etc. By doing so, one would expect to unleash the T cells, resulting in T cell proliferation, activation and a therapeutic response.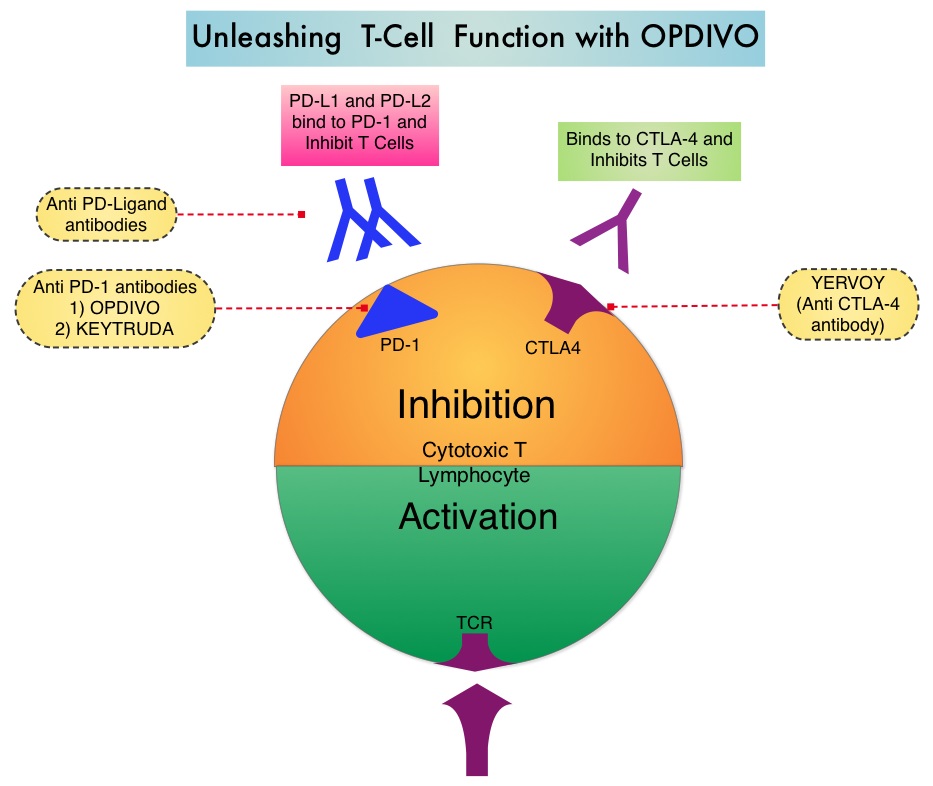
OPDIVO® is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response. CheckMate-141 is an open-label, phase III trial, in which 361 patients with recurrent or metastatic Squamous Cell Carcinoma of the oral cavity, pharynx, or larynx, were randomly assigned in a 2:1 ratio to receive either OPDIVO® (N= 240) or investigator’s choice of ERBITUX® (Cetuximab), TAXOTERE® (Docetaxel) or Methotrexate (N=121). OPDIVO® was administered intravenously at 3 mg/kg every 2 weeks. ERBITUX® dosing was 400 mg/m2 for the first dose followed by 250 mg/m2 weekly, TAXOTERE® dosing was 30 mg/m2 weekly and Methotrexate dosing was 40 mg/m2 weekly. The median age was 60 years, majority of patients were male, had received 2 or more prior systemic therapies and prior radiation therapy. Patients were stratified based on prior therapy with monoclonal antibody, ERBITUX® and HPV status was known for 50% of the participants. The primary endpoint was Overall Survival and secondary endpoints included Response Rates and Progression Free Survival (PFS).
This study was stopped earlier than scheduled after an independent monitoring panel determined that the primary endpoint for this study was met, with significant superiority of OPDIVO® over the investigator’s choice of therapy. The median Overall Survival with OPDIVO® was 7.5 months, compared to 5.1 months with investigator’s choice of therapy (HR=0.70; P=0.010). The 1-year Overall Survival with OPDIVO® was 36%, which was more than double, compared with investigator’s choice of therapy, which was 16.6%. Patients with PD-L1 expression of 1% or more benefited the most from treatment with OPDIVO®, with a median OS of 8.7 months, compared to 4.6 months in the control group (HR=0.55). For patients with PD-L1 expression less than 1%, median OS was 5.7 with OPDIVO® versus 5.8 months, for the control group (HR=0.89). HPV status also had an impact on outcomes. HPV-positive patients had a median OS of 9.1 months with OPDIVO® compared with 4.4 months with investigator’s choice of therapy (HR=0.56), whereas those who were HPV-negative and receiving OPDIVO®, had a median OS of 7.5 months compared to 5.8 months with investigator’s choice of therapy (HR=0.73). Grade 3 or 4 adverse events were significantly lower with OPDIVO®, compared with investigator’s choice of therapy(13.1% vs 35.1%).
The authors concluded that OPDIVO® is the first agent to demonstrate a significant improvement in Overall Survival, in patients with advanced head and neck cancer following progression on platinum-based chemotherapy, and fulfills an unmet need for this patient group. Gillison ML, Blumenschein G, Fayette J, et al. Nivolumab Versus Investigator’s Choice (IC) for Recurrent or Metastatic (R/M) Head and Neck Squamous Cell Carcinoma (SCCHN): CheckMate-141. Presented at: AACR 2016 Annual Meeting, New Orleans; April 16-20, 2016. Abstract CT099.
Left Sided Colon Cancer Patients Have Better Outcomes than Those with Right Sided Tumors
SUMMARY: ColoRectal Cancer (CRC) is the third most common cancer diagnosed in both men and women in the United States. The American Cancer Society estimates that approximately 135,000 new cases of ColoRectal Cancer will be diagnosed in the United States in 2016 and over 49,000 patients are expected to die of the disease. Even though prognosis for patients with Colon Cancer has improved over the past two to three decades, it has remained unclear if improvement in survival was greater for Left sided Colon Cancer versus cancer originating in the Right hemicolon. To address this question, the authors in this study used the Geneva population-based registry and collected the data on all Colon Cancer patients from 1980-2006. They then compared outcomes ided Tumorsin 3396 patients with proximal (Right-sided) and distal (Left-sided) Colon Cancer. In the study population, 1,334 patients (39%) had Right-sided tumors and 2,062 (61%) had Left-sided tumors. Colon Cancer specific survival was then determined and compared, taking into consideration tumor and treatment characteristics of Left and Right sided tumors, as well as putative confounding variables. The authors also compared changes in survival between Colon Cancer location in early and late years of this observation study.
At the beginning of the study period (1980s), the 5-year specific survival was identical for Right and Left sided Colon Cancers (49% versus 48%). However, over the course of the study period, there was a marked improvement in survival, for patients with Left-sided cancers (HR=0.42; P<0.001), but this benefit was not noted in Right-sided Colon Cancer patients (HR=0.76; P=0.69). As such, distal Colon Cancer (Left sided) had a better prognosis than patients with proximal or Right sided Colon Cancer (Hazard Ratio for Left versus Right Colon Cancer = 0.81; P<0.001).
The authors concluded from this study that survival of patients with Right Colon Cancer did not improve since 1980, and tend to have the worse prognosis of all Colon Cancer patients. Therefore, change in treatment strategy is warranted for this patient subgroup. Right colon cancer: left behind. Gervaza P, Usel M, Rapiti E, et al. European Journal of Surgical Oncology 2016;doi:10.1016/j.ejso.2016.04.002