SUMMARY: A Biosimilar product is a biological product that is approved based on its high similarity to an already approved biological product (also known as reference product). Biological products are made from living organisms including humans, animals and microorganisms such as bacteria or yeast and are manufactured through biotechnology, derived from natural sources or produced synthetically. Biological products have larger molecules with a complex structure than conventional drugs (also known as small molecule drugs). Unlike biological products, conventional drugs are made of pure chemical substances and their structures can be identified. A generic drug is a copy of brand name drug and has the same active ingredient and is the same as brand name drug in dosage form, safety and strength, route of administration, quality, performance characteristics and intended use. Therefore, brand name and the generic drugs are bioequivalent. 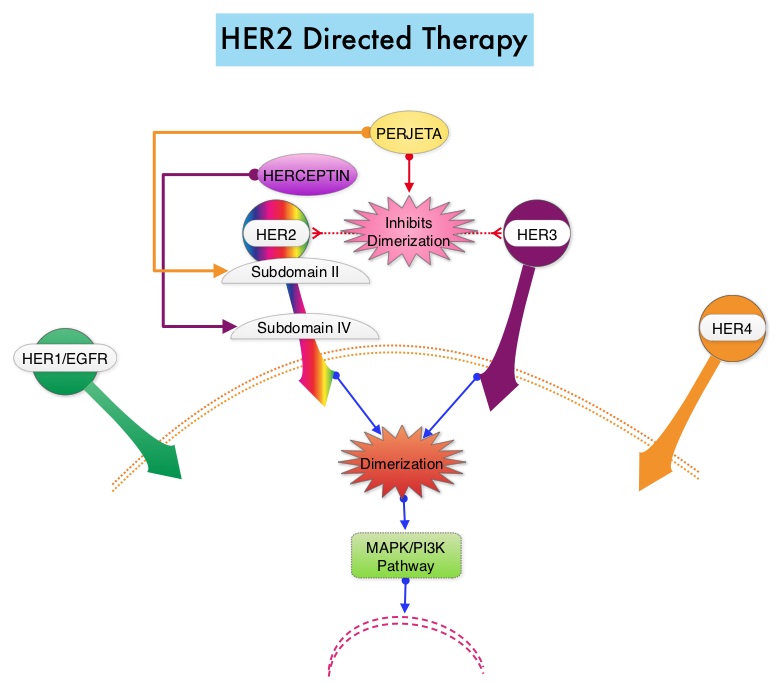 The Affordable Care Act in 2010 created an abbreviated licensure pathway for biological products that are demonstrated to be “Biosimilar” to, or “interchangeable” with an FDA-licensed (FDA approved) biological product (reference product). The Biosimilar must show that it has no clinically meaningful differences in terms of safety and effectiveness from the reference product. A Biosimilar product can only be approved by the FDA if it has the same mechanism of action, route of administration, dosage form and strength as the reference product, and only for the indications and conditions of use that have been approved for the reference product. Biosimilars are not as easy to manufacture as generics (copies of brand name drugs) because of the complexity of the structure of the biologic product and the process used to make a biologic product. The facilities where Biosimilars are manufactured must also meet the FDA’s standards.
The Affordable Care Act in 2010 created an abbreviated licensure pathway for biological products that are demonstrated to be “Biosimilar” to, or “interchangeable” with an FDA-licensed (FDA approved) biological product (reference product). The Biosimilar must show that it has no clinically meaningful differences in terms of safety and effectiveness from the reference product. A Biosimilar product can only be approved by the FDA if it has the same mechanism of action, route of administration, dosage form and strength as the reference product, and only for the indications and conditions of use that have been approved for the reference product. Biosimilars are not as easy to manufacture as generics (copies of brand name drugs) because of the complexity of the structure of the biologic product and the process used to make a biologic product. The facilities where Biosimilars are manufactured must also meet the FDA’s standards.
Heritage is a double-blind, randomized phase III trial in which the efficacy and safety of Myl-1401O, a Biosimilar, was compared with HERCEPTIN®. The randomization included 500 patients treated at 95 sites worldwide, with centrally confirmed, measurable HER2 positive metastatic breast cancer, who had not received prior chemotherapy or HERCEPTIN® for their metastatic disease. Patients received either Myl-1401O or HERCEPTIN® along with TAXOTERE® (Docetaxel) or TAXOL® (Paclitaxel) administered every 3 weeks for a minimum of 8 cycles (24 weeks), with the antibody therapy continued, until disease progression. Both antibodies were administered with a loading dose of 8 mg/kg and a maintenance dose of 6 mg/kg every 3 weeks. Approximately 44% of the enrolled patients had hormone receptor positive disease and 84% received TAXOTERE®. The final analysis included 458 patients of whom 230 were in the Myl-1401O group and 228 were in the HERCEPTIN® group. The Primary endpoint was Overall Response Rate (ORR) at 24 weeks and Secondary endpoints include Progression Free Survival (PFS), Overall Survival (OS) and Safety.
The ORR after 24 weeks of treatment was 69.6% for the Myl-1401O group and 64% for the HERCEPTIN® group and this was not statistically significant. The median PFS had not yet been reached. Safety data in both treatment groups were comparable and there was no significant change in cardiac function from baseline to Week 24 in either group. The dose-normalized maximum concentration and Area Under the Curve, were similar for both antibodies.
The authors concluded that this study is one of the first trials of Biosimilars in oncology to demonstrate similar results and they added that MYL-1401O is equivalent to HERCEPTIN®, when given in combination with a Taxane, as first line therapy, for patients with HER2 positive metastatic breast cancer. Heritage: A phase III safety and efficacy trial of the proposed trastuzumab biosimilar Myl-1401O versus Herceptin. Rugo HS, Barve A, Waller CF, et al. J Clin Oncol 34, 2016 (suppl; abstr LBA503)
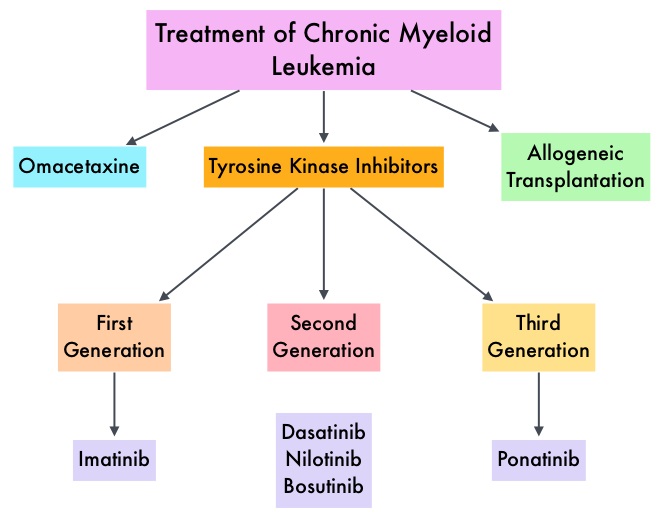 The presently available Tyrosine Kinase Inhibitors (TKI’s) approved in the United States including GLEEVEC®, share the same therapeutic target, which is BCR-ABL kinase. Resistance to TKI’s can occur as a result of mutations in the BCR-ABL kinase domain or amplification of the BCR-ABL gene. With the availability of newer therapies for CML, monitoring response to treatment is important. This is best accomplished by measuring the amount of residual disease using Reverse Transcription-Polymerase Chain Reaction (RT-PCR). Molecular response in CML is expressed using the International Scale (IS) as BCR-ABL%, which is the ratio between BCR-ABL and a control gene. BCR-ABL kinase domain point mutations are detected, using the mutational analysis by Sanger sequencing. Majority of the patients receiving a TKI following diagnosis of CML achieve a Complete Cytogenetic Response (CCyR) within 12 months following commencement of therapy and these patients have a life expectancy similar to that of their healthy counterparts. Previously published studies have shown that deep molecular response (MR4.5) is a new molecular predictor of long term survival in CML patients and was achieved in a majority of patients treated with optimized dose of GLEEVEC®. It has been hypothesized based on previous observations, that a subgroup of CML patients experiencing deeper responses (MR3, MR4, and MR4.5), may stay in unmaintained remission even after treatment discontinuation. Despite this observation, stopping CML therapy is currently not a clinical recommendation and should only be considered in the context of a clinical trial.
The presently available Tyrosine Kinase Inhibitors (TKI’s) approved in the United States including GLEEVEC®, share the same therapeutic target, which is BCR-ABL kinase. Resistance to TKI’s can occur as a result of mutations in the BCR-ABL kinase domain or amplification of the BCR-ABL gene. With the availability of newer therapies for CML, monitoring response to treatment is important. This is best accomplished by measuring the amount of residual disease using Reverse Transcription-Polymerase Chain Reaction (RT-PCR). Molecular response in CML is expressed using the International Scale (IS) as BCR-ABL%, which is the ratio between BCR-ABL and a control gene. BCR-ABL kinase domain point mutations are detected, using the mutational analysis by Sanger sequencing. Majority of the patients receiving a TKI following diagnosis of CML achieve a Complete Cytogenetic Response (CCyR) within 12 months following commencement of therapy and these patients have a life expectancy similar to that of their healthy counterparts. Previously published studies have shown that deep molecular response (MR4.5) is a new molecular predictor of long term survival in CML patients and was achieved in a majority of patients treated with optimized dose of GLEEVEC®. It has been hypothesized based on previous observations, that a subgroup of CML patients experiencing deeper responses (MR3, MR4, and MR4.5), may stay in unmaintained remission even after treatment discontinuation. Despite this observation, stopping CML therapy is currently not a clinical recommendation and should only be considered in the context of a clinical trial.
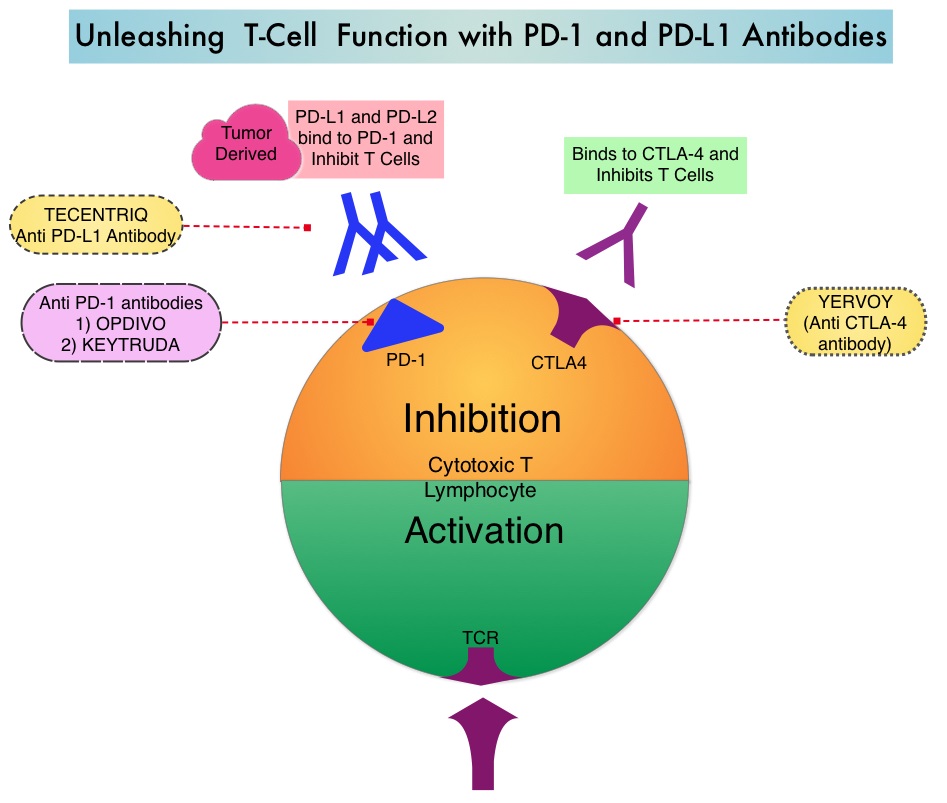 The treatment paradigm for solid tumors has been rapidly evolving with a better understanding of the Immune checkpoints or gate keepers. Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. Survival of cancer cells in the human body may be to a significant extent related to their ability to escape immune surveillance by inhibiting T lymphocyte activation. The T cells of the immune system therefore play a very important role in modulating the immune system. Under normal circumstances, inhibition of an intense immune response and switching off the T cells of the immune system, is an evolutionary mechanism and is accomplished by Immune checkpoints or gate keepers. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies are being developed that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152), PD-1(Programmed cell Death 1), etc. By doing so, one would expect to unleash the T cells, resulting in T cell proliferation, activation and a therapeutic response.
The treatment paradigm for solid tumors has been rapidly evolving with a better understanding of the Immune checkpoints or gate keepers. Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. Survival of cancer cells in the human body may be to a significant extent related to their ability to escape immune surveillance by inhibiting T lymphocyte activation. The T cells of the immune system therefore play a very important role in modulating the immune system. Under normal circumstances, inhibition of an intense immune response and switching off the T cells of the immune system, is an evolutionary mechanism and is accomplished by Immune checkpoints or gate keepers. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies are being developed that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152), PD-1(Programmed cell Death 1), etc. By doing so, one would expect to unleash the T cells, resulting in T cell proliferation, activation and a therapeutic response.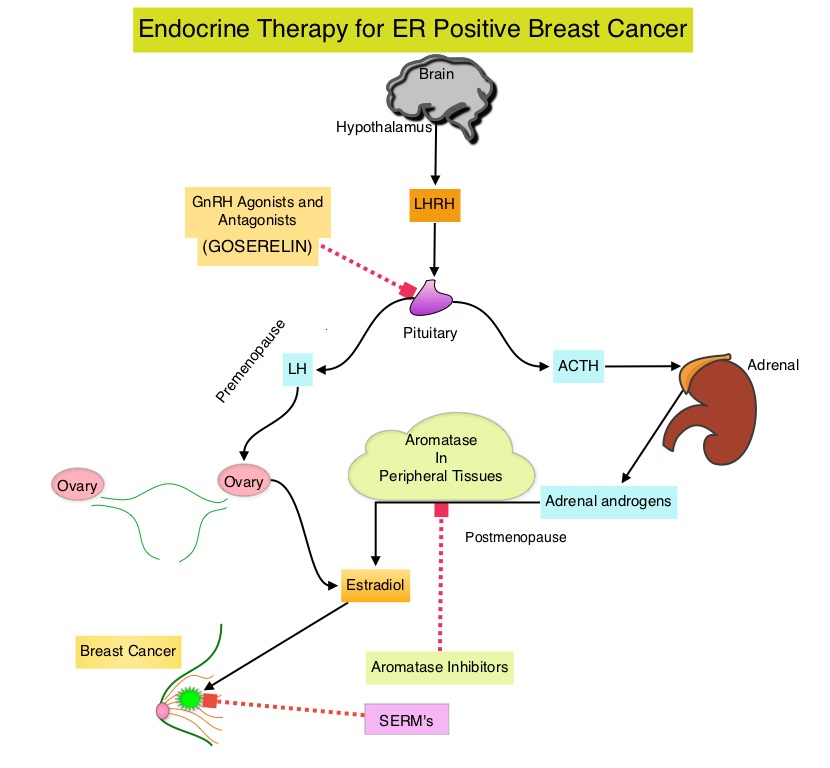 These patients are often treated with anti-estrogen therapy as first line treatment. In premenopausal woman, the ovary is the main source of estrogen production, whereas in postmenopausal women, the primary source of estrogen is the Aromatase enzyme mediated conversion of androstenedione and testosterone to estrone and estradiol, in extragonadal/peripheral tissues. NOLVADEX® (Tamoxifen) is a nonsteroidal Selective Estrogen Receptor Modulator (SERM) and works mainly by binding to the Estrogen Receptor and thus blocks the proliferative actions of estrogen on the mammary tissue. ARIMIDEX® (Anastrozole), FEMARA® (Letrozole) and AROMASIN® (Exemestane) are Aromatase Inhibitors (AIs) that binds to the Aromatase enzyme and inhibit the conversion of androgens to estrogens in the extra-gonadal tissues.
These patients are often treated with anti-estrogen therapy as first line treatment. In premenopausal woman, the ovary is the main source of estrogen production, whereas in postmenopausal women, the primary source of estrogen is the Aromatase enzyme mediated conversion of androstenedione and testosterone to estrone and estradiol, in extragonadal/peripheral tissues. NOLVADEX® (Tamoxifen) is a nonsteroidal Selective Estrogen Receptor Modulator (SERM) and works mainly by binding to the Estrogen Receptor and thus blocks the proliferative actions of estrogen on the mammary tissue. ARIMIDEX® (Anastrozole), FEMARA® (Letrozole) and AROMASIN® (Exemestane) are Aromatase Inhibitors (AIs) that binds to the Aromatase enzyme and inhibit the conversion of androgens to estrogens in the extra-gonadal tissues.