SUMMARY: There are presently four New Oral Anticoagulants approved in the United States for the treatment of Venous ThromboEmbolism. They include PRADAXA® (Dabigatran), which is a direct thrombin inhibitor and XARELTO® (Rivaroxaban), ELIQUIS® (Apixaban), SAVAYSA® (Endoxaban), which are Factor Xa inhibitors. Compared to COUMADIN® (Warfarin), the New Oral Anticoagulants have a rapid onset of action, wider therapeutic window, shorter half-lives (7-14 hours in healthy individuals), no laboratory monitoring and fixed dosing schedule. The half life of these agents can however be prolonged in those with renal insufficiency and may be unsafe and direct oral anticoagulants are ineffective in patients with mechanical heart valves. In several clinical studies, these New Oral Anticoagulants have been shown to reduce the rate of major bleeding by 28% and the rates of intracranial and fatal hemorrhage by 50%, when compared to COUMADIN®. Unlike bleeding caused by COUMADIN®, which can be reversed using Vitamin K or Fresh Frozen Plasma, there are no specific agents presently available, for reversing bleeding caused by the New Oral Anticoagulants or for stopping the anticoagulant effects of these drugs, in patients who need urgent surgical intervention. The FDA in October 16, 2015, granted accelerated approval to Idarucizumab (PRAXBIND®), for the treatment of patients treated with Dabigatran (PRADAXA®), a direct thrombin inhibitor, when reversal of the anticoagulant effects of PRADAXA® is needed for emergency surgery/urgent procedures, or in life-threatening or uncontrolled bleeding. However, the other New Oral Anticoagulants approved in the United States for the treatment of Venous ThromboEmbolism such as XARELTO® (Rivaroxaban), ELIQUIS® (Apixaban), SAVAYSA® (Endoxaban), are Factor Xa inhibitors and do not have an antidote. As such, some Health Care Providers have discouraged their patients from taking these direct oral anticoagulants until an antidote became available, should their patients need urgent surgical intervention.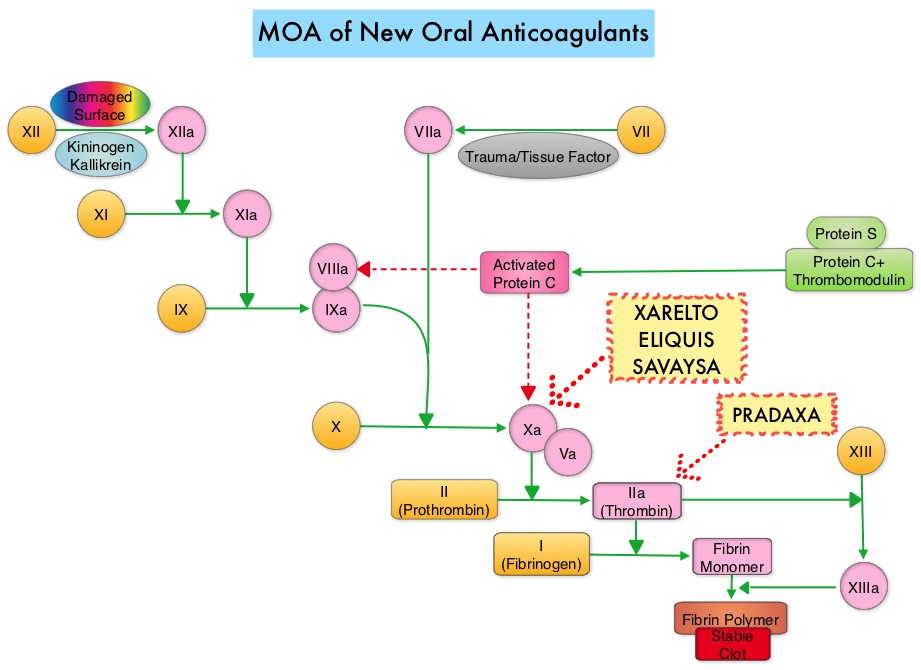
Andexanet alfa (AndexXa®) is a recombinant, modified human Factor Xa decoy protein without intrinsic catalytic activity, that binds Factor Xa inhibitors. In a previously published study, AndexXa® reversed the anticoagulant activity of ELIQUIS® and XARELTO® in older healthy participants within minutes after administration, and reduced both the unbound fraction of the plasma level of factor Xa inhibitor and anti-Factor Xa activity, with minimal clinical toxicity (N Engl J Med 2015; 373:2413-242). The AndexXa®, a Novel Antidote to the Anticoagulation Effects of FXA Inhibitors (ANNEXA-4) study is an ongoing, multicenter, prospective, open-label, single-group study designed to evaluate the use of AndexXa® in patients with acute major bleeding that was potentially life-threatening.
The authors in this interim report described the outcomes of 67 patients, for whom complete data was available. Patients in this study had acute major bleeding within 18 hours after the administration of one of four Factor Xa inhibitors – ELIQUIS®, XARELTO®, SAVAYSA®, or LOVENOX® (Enoxaparin). Acute major bleeding was defined as potentially life-threatening with signs or symptoms of hemodynamic compromise, decrease in hemoglobin of at least 2 gm/dl, symptomatic bleeding in a critical area or organ. The mean patient age was 77 years, and most patients had a history of cardiovascular disease and thrombotic events and bleeding was predominantly gastrointestinal or intracranial. An independent committee determined hemostatic efficacy on the basis of predetermined criteria. The baseline value for anti-Factor Xa activity was at least 75 ng/ml and 0.5 IU/ml or more for those receiving LOVENOX® and patients had confirmed bleeding severity. All patients received a bolus dose of AndexXa® within 3-6 hours following presentation to the ER followed by a 2-hour infusion of the drug. The two co-primary outcomes were the percent change in the anti-Factor Xa activity and the rate of excellent or good hemostatic efficacy, 12 hours after the AndexXa® infusion. Anti-Factor Xa activity was measured by means of a validated chromogenic assay of Factor Xa enzymatic activity.
It was noted that following the bolus dose of AndexXa®, the median anti-Factor Xa activity decreased by 89% from baseline, among patients receiving XARELTO® and by 93% among patients receiving ELIQUIS® and these levels remained the same during the 2-hour infusion. Four hours after the end of the infusion, the relative decrease in the anti-Factor Xa activity from baseline, among patients receiving XARELTO® was 39% and 30% among those receiving ELIQUIS®. Twelve hours after the AndexXa® infusion, hemostatic efficacy was adjudicated as excellent or good by the independent committee, in 79% of the patients and was consistent across all subgroups. During the 30-day follow up period, thrombotic events occurred in 18% of the patients.
The authors concluded that AndexXa® rapidly reversed anti-Factor Xa activity without significant toxicity, in 79% of the patients, 12 hours after an infusion of AndexXa®. This study also demonstrated that prolonged reversal of Factor Xa inhibition may not be necessary to achieve a good hemostatic response with AndexXa®. Andexanet Alfa for Acute Major Bleeding Associated with Factor Xa Inhibitors. Connolly SJ, Milling TJ, Eikelboom JW, et al. N Engl J Med 2016; 375:1131-1141
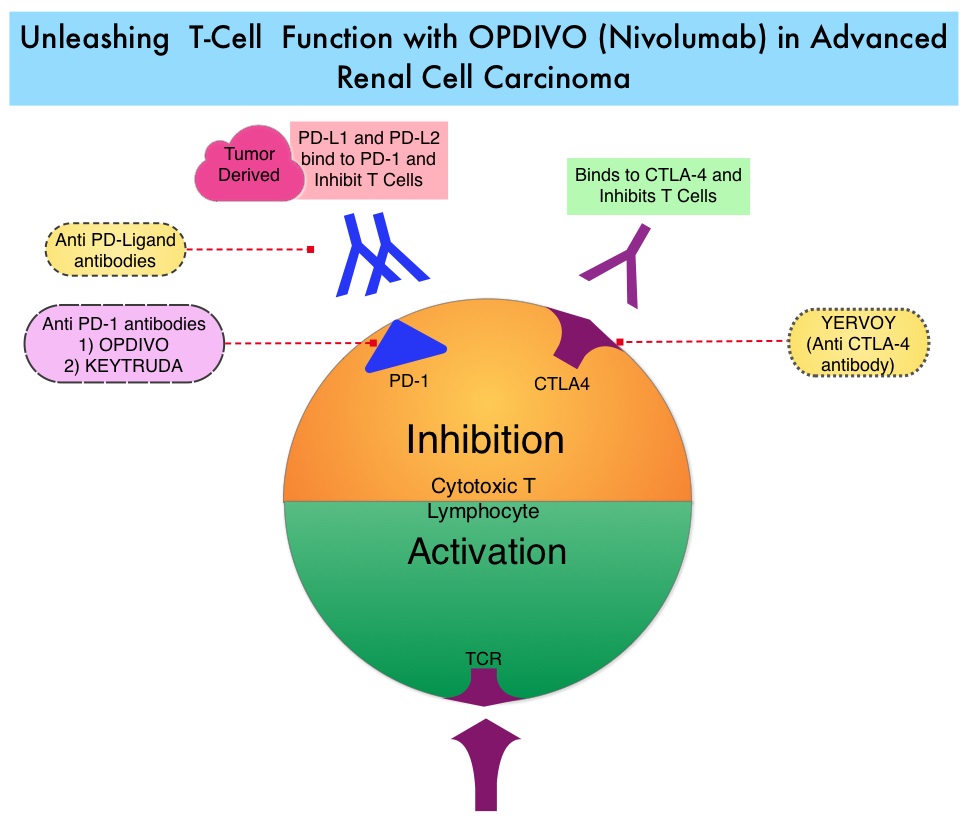
 In the phase II study, 167 patients with advanced Renal Cell Carcinoma with KPS of 70% or more and who had prior treatment with 1-3 regimens in the metastatic setting, received OPDIVO® at a dose of 0.3, 2, or 10 mg/kg every 3 weeks (J Clin Oncol 2015;33:1430–37). At a minimum follow up of 38 months, ORR was 21% and the median Duration of Response was 22 months. The 3 year OS rate was 35% and 4 year OS rate was 29%. Long-term survival was observed in MSKCC good, intermediate and poor-risk patients, as well as in patients with excellent or reduced Karnofsky Performance Status.
In the phase II study, 167 patients with advanced Renal Cell Carcinoma with KPS of 70% or more and who had prior treatment with 1-3 regimens in the metastatic setting, received OPDIVO® at a dose of 0.3, 2, or 10 mg/kg every 3 weeks (J Clin Oncol 2015;33:1430–37). At a minimum follow up of 38 months, ORR was 21% and the median Duration of Response was 22 months. The 3 year OS rate was 35% and 4 year OS rate was 29%. Long-term survival was observed in MSKCC good, intermediate and poor-risk patients, as well as in patients with excellent or reduced Karnofsky Performance Status.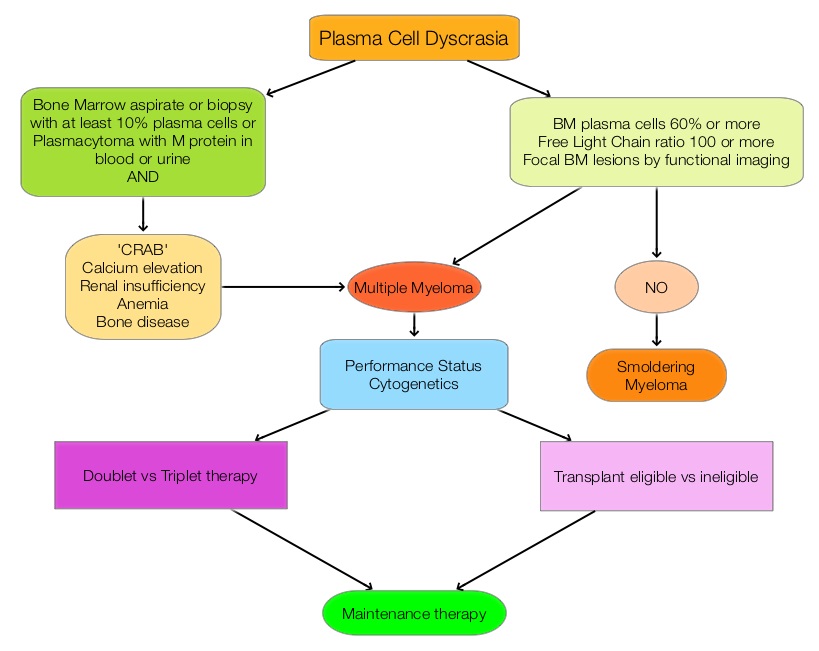 The recent new drugs approved for the treatment of relapsed/refractory Multiple Myeloma include a Histone Decetylase inhibitor (FARYDAK®) and 2 monoclonal antibodies, Daratumumab (DARZALEX®) and Elotuzumab (EMPLICITI®). The two most important determinants of Myeloma patient outcomes include, Performance Status and tumor genomics. Performance Status can be assessed using currently validated instruments and patients can be defined as Fit, Intermediate Fit, or Frail. Patients with chromosomal abnormalities t(4;14), t(14;16), t(14;20) or del 17p are considered to fall in the high risk group.
The recent new drugs approved for the treatment of relapsed/refractory Multiple Myeloma include a Histone Decetylase inhibitor (FARYDAK®) and 2 monoclonal antibodies, Daratumumab (DARZALEX®) and Elotuzumab (EMPLICITI®). The two most important determinants of Myeloma patient outcomes include, Performance Status and tumor genomics. Performance Status can be assessed using currently validated instruments and patients can be defined as Fit, Intermediate Fit, or Frail. Patients with chromosomal abnormalities t(4;14), t(14;16), t(14;20) or del 17p are considered to fall in the high risk group.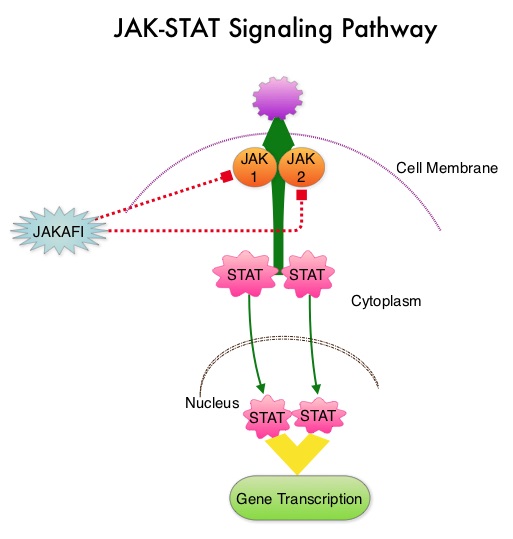 The JAK-STAT signaling pathway has been implicated in the pathogenesis of Myelofibrosis. This pathway normally is responsible for passing information from outside the cell through the cell membrane to the DNA in the nucleus for gene transcription. Janus Kinase (JAK) family of tyrosine kinases are cytoplasmic proteins and include JAK1, JAK2, JAK3 and TYK2. JAK1 helps propagate the signaling of inflammatory cytokines whereas JAK2 is essential for growth and differentiation of hematopoietic stem cells. These tyrosine kinases mediate cell signaling by recruiting STAT’s (Signal Transducer and Activator of Transcription), with resulting modulation of gene expression. In patients with MPN, the aberrant myeloproliferation is the result of dysregulated JAK2-STAT signaling as well as excess production of inflammatory cytokines associated with this abnormal signaling. These cytokines contribute to the symptoms often reported by patients with MF. JAK2 mutations such as JAK2 V617F are seen in approximately 60% of the patients with PMF and ET and 95% of patients with PV. Unlike CML where the BCR-ABL fusion gene triggers the disease, JAK2 mutations are not initiators of the disease and are not specific for MPN. Further, several other genetic events may contribute to the abnormal JAK2-STAT signaling.
The JAK-STAT signaling pathway has been implicated in the pathogenesis of Myelofibrosis. This pathway normally is responsible for passing information from outside the cell through the cell membrane to the DNA in the nucleus for gene transcription. Janus Kinase (JAK) family of tyrosine kinases are cytoplasmic proteins and include JAK1, JAK2, JAK3 and TYK2. JAK1 helps propagate the signaling of inflammatory cytokines whereas JAK2 is essential for growth and differentiation of hematopoietic stem cells. These tyrosine kinases mediate cell signaling by recruiting STAT’s (Signal Transducer and Activator of Transcription), with resulting modulation of gene expression. In patients with MPN, the aberrant myeloproliferation is the result of dysregulated JAK2-STAT signaling as well as excess production of inflammatory cytokines associated with this abnormal signaling. These cytokines contribute to the symptoms often reported by patients with MF. JAK2 mutations such as JAK2 V617F are seen in approximately 60% of the patients with PMF and ET and 95% of patients with PV. Unlike CML where the BCR-ABL fusion gene triggers the disease, JAK2 mutations are not initiators of the disease and are not specific for MPN. Further, several other genetic events may contribute to the abnormal JAK2-STAT signaling. The authors now reported the final long term efficacy and safety results after 5 years of treatment with JAKAFI® in the COMFORT-I study. In COMFORT-I study, 309 intermediate or high risk patients were randomized to receive either JAKAFI® (N=155) or Placebo (N=154). The Primary end point was a 35% or more reduction in spleen size at 24 weeks. The preplanned 5- year analysis occurred when all patients reached the 5-year visit or discontinued treatment. Patients in the placebo group could crossover to the JAKAFI® group after the primary analysis (when all patients completed week 24) or at any time if they had pre-specified worsening of splenomegaly. Of the 154 patients randomized to placebo, 111 patients crossed over to the JAKAFI® group and the median time to crossover was 41 weeks.
The authors now reported the final long term efficacy and safety results after 5 years of treatment with JAKAFI® in the COMFORT-I study. In COMFORT-I study, 309 intermediate or high risk patients were randomized to receive either JAKAFI® (N=155) or Placebo (N=154). The Primary end point was a 35% or more reduction in spleen size at 24 weeks. The preplanned 5- year analysis occurred when all patients reached the 5-year visit or discontinued treatment. Patients in the placebo group could crossover to the JAKAFI® group after the primary analysis (when all patients completed week 24) or at any time if they had pre-specified worsening of splenomegaly. Of the 154 patients randomized to placebo, 111 patients crossed over to the JAKAFI® group and the median time to crossover was 41 weeks.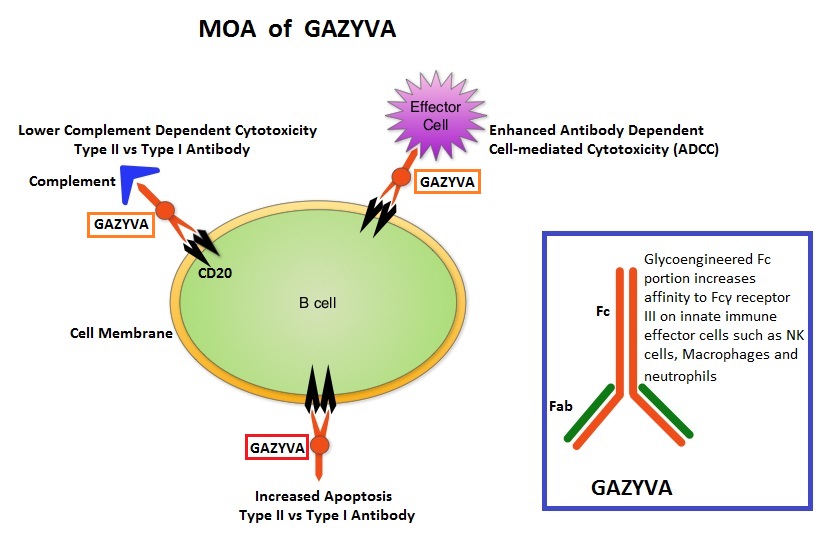 GAZYVA® (Obinutuzumab) is glycoengineered, fully humanized, third generation, type II anti-CD20 antibody (IgG1 monoclonal antibody) that selectivity binds to the extracellular domain of the CD20 antigen on malignant human B cells. By virtue of binding affinity of the glycoengineered Fc portion of GAZYVA® to Fcγ receptor III on innate immune effector cells such as natural killer cells, macrophages and neutrophils, Antibody-Dependent Cell-mediated Cytotoxicity (ADCC) and Antibody-Dependent Cellular phagocytosis is significantly enhanced, whereas it induces very little Complement-Dependent Cytotoxicity. This is in contrast to RITUXAN® (Rituximab), which is a first generation type I, chimeric anti-CD20 targeted monoclonal antibody that kills lymphoma cells primarily by Complement-Dependent Cytotoxicity and also ADCC.
GAZYVA® (Obinutuzumab) is glycoengineered, fully humanized, third generation, type II anti-CD20 antibody (IgG1 monoclonal antibody) that selectivity binds to the extracellular domain of the CD20 antigen on malignant human B cells. By virtue of binding affinity of the glycoengineered Fc portion of GAZYVA® to Fcγ receptor III on innate immune effector cells such as natural killer cells, macrophages and neutrophils, Antibody-Dependent Cell-mediated Cytotoxicity (ADCC) and Antibody-Dependent Cellular phagocytosis is significantly enhanced, whereas it induces very little Complement-Dependent Cytotoxicity. This is in contrast to RITUXAN® (Rituximab), which is a first generation type I, chimeric anti-CD20 targeted monoclonal antibody that kills lymphoma cells primarily by Complement-Dependent Cytotoxicity and also ADCC.