SUMMARY: The U.S. Food and Drug Administration on February 28, 2017 approved XERMELO® (Telotristat ethyl) tablets in combination with SomatoStatin Analog (SSA) therapy for the treatment of adults with Carcinoid syndrome diarrhea that SSA therapy alone has inadequately controlled. The hallmark of Carcinoid syndrome is excessive tumor derived serotonin secretion. Carcinoid syndrome is characterized by flushing, diarrhea, bronchial constriction, and the development of cardiac valvular fibrosis, which may lead to heart failure. Diarrhea in these patients can be debilitating with significant impact on quality of life. Serotonin is metabolized into 5-HydyroxyIndoleAcetic Acid (5-HIAA) which can be measured in the urine and is often used to follow response to treatment in patients with Carcinoid syndrome. Somatostatin analogs (SSAs) target somatostatin receptors and can regulate important cellular activity as well as help inhibit hypersecretion of hormones, including serotonin and growth hormone. This in turn can control severe diarrhea and flushing associated with metastatic Carcinoid tumors. SSAs are often used as initial treatment in this patient group, although patients may develop recurrent or progressive symptoms during the course of their disease. Tryptophan hydroxylase (TPH), the rate-limiting enzyme in serotonin synthesis, converts tryptophan to 5-hydroxytryptophan, which is then converted to serotonin.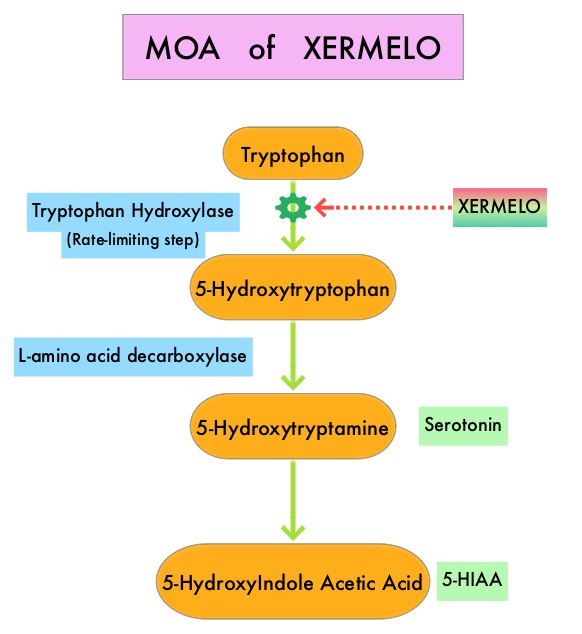
XERMELO® is a novel, oral, small-molecule TPH inhibitor, with a high molecular weight, that reduces peripheral serotonin production by inhibiting the enzyme TPH in metastatic carcinoid tumors. XERMELO® was shown in previously published studies, to reduce bowel movement (BM) frequency and decrease urinary 5-HIAA, in patients with Carcinoid syndrome, without causing CNS adverse effects.
TELESTAR is an international, multicenter, randomized, double-blind, placebo-controlled phase III trial in which the safety and efficacy of XERMELO® was evaluated in patients with Carcinoid syndrome not adequately controlled with SSA therapy. One hundred and thirty five (N=135) patients were randomly assigned in a 1:1:1 ratio to receive oral doses of XERMELO® 250 mg, XERMELO® 500 mg, or placebo respectively, three times a day for 12 weeks. Eligible patients experienced four or more BMs per day despite treatment with a stable dose of SomatoStatin Analog (SSA). All patients continued to receive their SSA therapy for the 12 week duration. Patients were allowed to receive rescue doses of short-acting Octreotide and antidiarrheal agents. Treatment groups were well balanced and the mean age was 64 years. The primary end point was mean reduction from baseline in daily Bowel Movements (BMs), averaged over 12 weeks. Secondary end points included change in the urinary 5-HIAA from baseline at week 12, the number of daily flushing episodes, and abdominal pain severity averaged over 12 weeks. In an open-label extension study, 115 patients subsequently continued to receive XERMELO® 500 mg three times a day.
It was noted that 33% of patients randomized to the XERMELO® group experienced an average reduction of two bowel movements per day compared to only 4% of patients randomized to the placebo group. At the week 12 analysis, both XERMELO® doses significantly reduced mean urinary 5-HydroxyIndole Acetic Acid compared to placebo (P<0.001). The most common adverse events in those receiving XERMELO® ,were nausea, headache, increased levels of the liver enzyme gamma-glutamyl transferase, peripheral edema and sometimes constipation. No new safety issues were noted, when patients were followed up during the open-label extension study and patients had sustained BM responses to treatment.
It was concluded that for patients with Carcinoid syndrome not adequately controlled by SomatoStatin Analogs (SSAs), addition of XERMELO® to SSA treatment was generally safe and well tolerated and resulted in significant reductions in BM frequency and urinary 5-HydroxyIndole Acetic Acid. Telotristat Ethyl, a Tryptophan Hydroxylase Inhibitor for the Treatment of Carcinoid Syndrome. Kulke MH, Hörsch D, Caplin ME, et al. DOI: http://dx.doi.org/10.1200/JCO.2016.69.2780