SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately, 255,180 new cases of invasive breast cancer will be diagnosed in 2017 and over 41,070 women will die of the disease. The HER or erbB family of receptors consist of HER1, HER2, HER3 and HER4. Approximately 15%-20% of invasive breast cancers overexpress HER2/neu oncogene, which is a negative predictor of outcomes without systemic therapy. HERCEPTIN® (Trastuzumab) is a humanized monoclonal antibody targeting HER2. Trastuzumab binds to subdomain IV of the HER2 extracellular domain and blocks the downstream cell signaling pathways (PI3K-AKT pathway) and induces Antibody Dependent Cellular Cytotoxicity (ADCC). Adjuvant chemotherapy given along with HERCEPTIN® reduces the risk of disease recurrence and death, among patients with HER2-positive early breast cancer.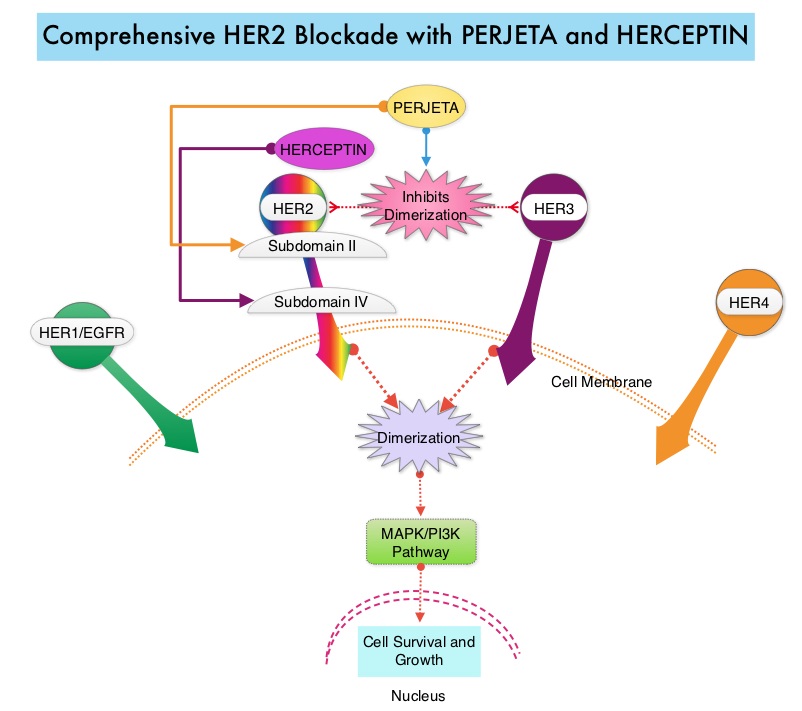
PERJETA® (Pertuzumab) is a recombinant humanized monoclonal antibody that binds to the HER2 at a different epitope of the HER2 extracellular domain (subdomain II) compared to HERCEPTIN® and prevents the dimerization of HER2 with HER3 receptor. PERJETA® induces ADCC similar to HERCEPTIN®. By combining HERCEPTIN® and PERJETA®, a more comprehensive blockade of HER2 signaling can be accomplished, as these two agents bind to different HER2 epitopes and may complement each other and improve efficacy. In the CLEOPATRA study, the addition of PERJETA® to HERCEPTIN® and Docetaxel resulted in significant improvement in Progression Free Survival (PFS) and Overall Survival (OS), in patients with HER-positive metastatic breast cancer. This triple drug combination also resulted in a significantly increased pathological Complete Response rate, when given in a neoadjuvant setting (NeoSphere trial).
Based on these previously published efficacy data, the authors in this study investigated whether the addition of PERJETA® to adjuvant HERCEPTIN® and chemotherapy, improves outcomes, among patients with HER2-positive early breast cancer. APHINITY is a prospective, randomized, multicenter, multinational, double-blind, placebo-controlled phase III trial in which a total of 4805 patients were randomly assigned in a 1:1 ratio, to receive standard adjuvant anthracycline or non-anthracycline chemotherapy regimen along with HERCEPTIN® plus either PERJETA® (2400 patients) or placebo (2405 patients). Anti-HER2 therapy was administered for a total of 1 year. Patients could receive radiotherapy and/or endocrine therapy following completion of adjuvant chemotherapy. Eligible patients had node-positive or high-risk node-negative (tumor diameter greater than 1.0 cm), HER2-positive, non-metastatic, adequately excised breast cancer. Both treatment groups were well balanced and about 37% of the patients had 1-3 positive lymph nodes and 25% of the patients had 4 or more positive lymph nodes. Two thirds of the patients were hormone receptor positive and about 78% of the patients received an anthracycline containing adjuvant chemotherapy regimen. The median follow up was 45.4 months and one year of treatment was completed by approximately 85% of the patients in both treatment groups. The primary end point was Disease Free Survival (DFS) from invasive breast cancer and secondary end points included Overall Survival (OS) and DFS from non-invasive breast cancers.
The addition of PERJETA® to chemotherapy and HERCEPTIN® resulted in a higher rate of DFS for invasive breast cancer with a 3-year invasive DFS of 94.1% in the PERJETA® group and 93.2% in the placebo group (HR=0.81; P=0.045), in favor of PERJETA®. Patients in the high risk subgroups benefited the most. The 3-year invasive DFS for patients with node-positive disease was 92.0% in the PERJETA® group, compared with 90.2% in the placebo group (HR=0.77; P=0.02). In the cohort of patients with hormone receptor negative tumors, the 3-year invasive DFS was 92.8% in the PERJETA® group and 91.2% in the placebo group (HR=0.76; P=0.08). The site of first distant recurrence was visceral or in the CNS rather than the bone. Cardiac toxicities were uncommon in both treatment groups and patients in the PERJETA® group had higher incidence of diarrhea while on concurrent chemotherapy.
It was concluded that for patients with HER2-positive early breast cancer, the addition of PERJETA® to standard postoperative HERCEPTIN® based adjuvant chemotherapy, significantly improved Disease Free Survival for invasive breast cancer. This benefit was more so for those patients with high risk disease. APHINITY trial (BIG 4-11): A randomized comparison of chemotherapy (C) plus trastuzumab (T) plus placebo (Pla) versus chemotherapy plus trastuzumab (T) plus pertuzumab (P) as adjuvant therapy in patients (pts) with HER2-positive early breast cancer (EBC). von Minckwitz G, Procter MJ, De Azambuja E, et al. J Clin Oncol. 2017;35(suppl; abstr LBA500).