
The FDA in September 2017, granted accelerated approval to ALIQOPA® (Copanlisib) for the treatment of adult patients with relapsed Follicular Lymphoma, who have received at least two prior systemic therapies. Follicular Lymphoma is the most indolent form and second most common form of all Non Hodgkin Lymphomas and approximately 30% of the patients will relapse in 3 years following initial treatment. ALIQOPA® is a pan-class 1, PI3K inhibitor with inhibitory activity predominantly against PI3K-α and PI3K-δ Isoforms expressed in malignant B cells. ALIQOPA® has significant activity in patients with relapsed/refractory indolent B-cell lymphoma, and the safety was manageable, compared with other PI3K inhibitors.
Month: October 2017
Circulating Tumor Cells in the Peripheral Blood May Predict Outcomes in Breast Cancer
SUMMARY: Circulating Tumor Cells (CTCs) are epithelial cells that are shed into the circulation from a primary or metastatic tumor. After being shed, CTCs can remain in the circulation or undergo apoptosis. Evaluation of CTCs during the course of disease has prognostic value. Because of the very low concentrations of CTCs (1 CTC in the background of millions of normal hematopoietic cells) in the peripheral blood, different technologies have been developed that will allow enrichment and detection of these CTCs. One such technology is the CellSearch® system which is the first FDA-approved test for CTC assessment, in the peripheral blood of patients with breast cancer. This automated system is able to enrich the peripheral blood sample with CTCs and the cells then are fluorescently stained for CytoKeratins (CK8,18 and 19), Common Leukocyte Antigen (CD45) and a nuclear dye (DAPI). CTCs are identified when they are CK positive, CD45 negative and DAPI positive. In essence, CTC assessment is a real time, peripheral blood evaluation (“Liquid Biopsy”) in breast cancer patients.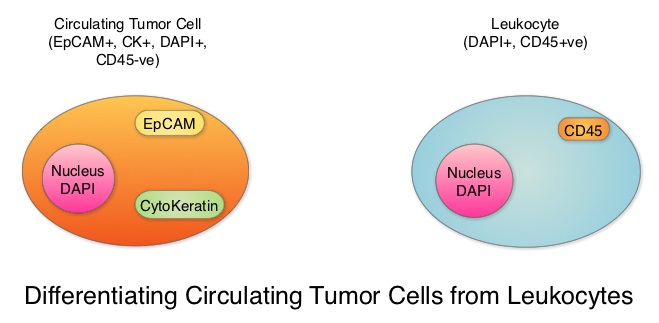
Despite advances in treatment, approximately 30% of patients with node-negative breast cancer and 50% of patients with node-positive breast cancer may relapse within 5 years. This is due to cancer cells shed from primary tumors that migrate to distal sites as Circulating Tumor Cells (CTCs) via the circulatory system. CTCs are therefore precursors of metastatic disease and may not only predict risk of metastatic disease but may also be useful in monitoring treatment efficacy. There have been conflicting reports about the effectiveness of different treatments to reduce CTCs in breast cancer patients, including the different molecular subtypes.
To further address these controversies, the authors conducted a meta-analysis of the published studies which included measurement of CTCs before and after treatment in breast cancer patients, and estimated the benefit of reducing CTC on patient outcomes. Data base searches included 1004 publications and 50 studies conducted between 2009 and 2016 in the US, Europe and Asia. A total of 6712 patients from these studies were eligible for meta-analysis. Enrolled patients had pathologically diagnosed breast cancer, CTCs were detected by any method, including cell capture and quantitative PCR and the patient’s CTC status both pre- and post-therapy was available. The CTC-positive rate was reported using different cut-off values of CTC count and different expression thresholds of epithelial genes (EpCAM, CK18, CK19) using RT-PCR in the various studies.
An overall analysis of the 6712 patients with CTC-positive rate by the random-effects model suggested that treatment intervention significantly decreased CTC-positive rate compared to the baseline (Relative Risk (RR)=0.68, P<0.00001, which meant a 32% reduction in the Relative Risk, compared to baseline values.
Subgroup analyses revealed that when compared to pre-treatment, CTCs were decreased after neoadjuvant treatment (RR=0.65, P=0.006), adjuvant treatment (RR=0.89, P=0.10), treatment in metastatic setting (RR=0.59, P<0.00001) and the combination therapy (RR=0.78, P=0.03). Reduction in CTCs was not seen after surgery (RR=1.27, P=0.42), suggesting that local intervention with surgery does not eliminate CTCs and patients with positive CTCs should receive other therapies after surgery, to decrease the risk of recurrence.
When compared to pre-treatment levels, treatment resulted in significant reduction in CTCs in HER2-positive patients (RR=0.68, P<0.0001) and HER2-negative patients (RR=0.52, P=0.01). This reduction in CTCs was however not noted in patients with triple-negative breast cancer patients (RR=0.38, P=0.29), indicating that current therapies for this group is inadequate and should be further optimized with newer therapies.
More importantly, reduction in CTCs was associated with lower probability of disease progression (P=0.01), longer Progression Free Survival (P<0.0001) and longer Overall Survival (P<0.00001).
It was concluded that based on this large meta-analysis, CTCs can help monitor the effectiveness of treatment and guide subsequent therapies in breast cancer patients. Circulating tumor cell status monitors the treatment responses in breast cancer patients: a meta-analysis. Yan W-T, Cui X, Chen Q, et al. Sci. Rep. 7, 43464; doi: 10.1038/srep43464 (2017).
FDA Approves CAR T-Cell Therapy for Non Hodgkin Lymphoma
SUMMARY: The FDA on October 18, 2017, granted regular approval to Axicabtagene ciloleucel (YESCARTA®) for the treatment of adult patients with relapsed or refractory Large B-Cell Lymphoma after two or more lines of systemic therapy, including Diffuse Large B-Cell Lymphoma (DLBCL) Not Otherwise Specified, Primary Mediastinal Large B-Cell Lymphoma, High-grade B-Cell Lymphoma, and DLBCL arising from Follicular Lymphoma (Transformed Follicular Lymphoma-TFL).
What is (CAR) T-cell immunotherapy? Chimeric Antigen Receptor (CAR) T-cell therapy is a type of immunotherapy and consists of T cells collected from the patient’s blood in a leukapheresis procedure, and genetically engineered to produce special receptors on their surface called Chimeric Antigen Receptors (CAR). These reprogrammed cytotoxic T cells with the Chimeric Antigen Receptors on their surface are now able to recognize a specific antigen on tumor cells. These genetically engineered and reprogrammed CAR T-cells are grown in the lab and are then infused into the patient. These cells in turn proliferate in the patient’s body and the engineered receptor on the cell surface help recognize and kill cancer cells that expresses that specific antigen. It is a therefore a customized treatment created using patient’s own T cells to destroy cancer cells.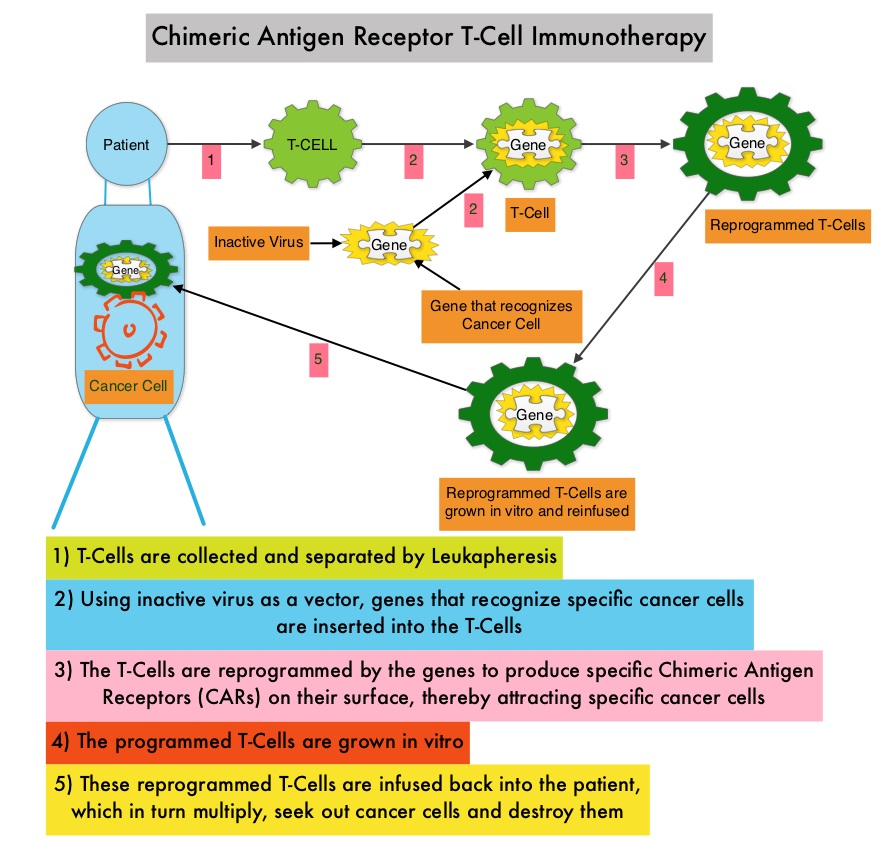
YESCARTA® is a Chimeric Antigen Receptor (CAR) T cell immunotherapy and consists of autologous T cells that are genetically modified to produce a CAR protein, allowing the T cells to seek out and destroy cancer cells expressing the antigen CD19, which is found uniquely on B cells. Patients, following treatment with CAR T-cells, develop B-cell aplasia (absence of CD19 positive cells) due to B-cell destruction and may need immunoglobin replacement. Hence, B-cell aplasia can be a useful therapeutic marker, as continued B-cell aplasia has been seen in all patients who had sustained remission, following CAR T-cell therapy. Cytokine Release Syndrome (CRS), an inflammatory process is the most common and serious side effect of CAR T-cell therapy and is associated with marked elevation of Interleukin-6. Cytokine release is important for T-cell activation and can result in high fevers and myalgias. This is usually self limiting although if severe can be associated with hypotension and respiratory insufficiency. Tocilizumab (ACTEMRA®), an Interleukin-6 receptor blocking antibody produces a rapid improvement in symptoms. This is however not recommended unless the symptoms are severe and life threatening, as blunting the cytokine response can in turn negate T-cell proliferation. Elevated serum Ferritin and C-reactive protein levels are surrogate markers for severe Cytokine Release Syndrome. The CAR T-cells have been shown to also access sanctuary sites such as the central nervous system and eradicate cancer cells. CD19 antigen is expressed by majority of the B cell malignancies and therefore most studies using CAR T-cell therapy have focused on the treatment of advanced B-cell malignancies such as Chronic Lymphocytic Leukemia (CLL), Acute Lymphoblastic Leukemia (ALL) and Non Hodgkin lymphoma (NHL), such as Diffuse Large B-Cell Lymphoma (DLBCL).
Diffuse Large B-Cell Lymphoma (DLBCL) is the most common of the aggressive Non-Hodgkin lymphoma’s in the United States, and the incidence has steadily increased 3 to 4% each year. Outcomes for patients with relapsed/refractory disease is poor, with an Objective response Rate (ORR) of 26%, Complete Response (CR) rate of 8% and a median Overall Survival (OS) of 6.6 months. There is therefore a significant unmet need in this patient group.
The safety and efficacy of YESCARTA® was evaluated in a single arm multicenter clinical trial (ZUMA-1 study) in which 111 patients were enrolled and 101 patients received therapy with YESCARTA®. This study included patients with DLBCL (N=77) and Primary Mediastinal B-Cell Lymphoma (PMBCL)/Transformed Follicular Lymphoma-TFL (N=24), with chemo refractory disease or patients who had relapsed within 12 months post Autologous Stem Cell Transplantation. Following 3 days of conditioning regimen with Cyclophosphamide and Fludarabine, patients received a single infusion of YESCARTA® at a dose of 2 x 106 CAR-positive T cells/kg. The average turnaround time from apheresis to delivery to clinical site for infusion, was 17 days. The Primary end point was Objective Response Rate (ORR) and Secondary endpoints included Duration of Response, Overall Survival and Safety. The preliminary results were reported after the Primary end point was met.
After a median follow up of 8.7 months, the ORR for the entire group was 82% with 54% CR rate (P<0.0001). Among those with DLBCL, the ORR was 82% and the CR rate was 49% and in the PMBCL/TFL group, the ORR was 83% and the CR rate was 71%. At the time of median follow up, 44% of the patients had ongoing responses and the median Duration of Response was 8.2 months and not reached for those in CR. The median Progression Free Survival was 5.9 months and median OS was not reached.
The most common grade 3 or higher adverse reactions included cytopenias, febrile neutropenia, fever, Cytokine Release Syndrome (CRS) and neurologic events. CRS and neurologic events were generally reversible and 43% received Tocilizumb and 27% received steroids and this did not negatively impact outcomes.
The authors concluded that ZUMA-1 is the first pivotal trial of CD19-specific CAR T-cell therapy in patients with refractory aggressive Non Hodgkin Lymphoma, demonstrating significant clinical activity, with a Complete Response rate 7 times higher than the historic control rate. Primary results from ZUMA-1: a pivotal trial of axicabtagene ciloleucel (Axi-cel; KTE-C19) in patients with refractory aggressive non-Hodgkin lymphoma (NHL). Locke FL, Neelapu SS, Bartlett NL, et al. Presented at: 2017 AACR Annual Meeting; April 1-5, 2017; Washington, DC. Abstract CT019.
GAZYVA® Superior to RITUXAN® for First-Line Treatment of Follicular Lymphoma
SUMMARY: The American Cancer Society estimates that in 2017, about 72,240 people will be diagnosed with Non Hodgkin Lymphoma (NHL) in the United States and about 20,140 individuals will die of this disease. Indolent Non Hodgkin Lymphomas are mature B cell lymphoproliferative disorders and include Follicular Lymphoma, Nodal Marginal Zone Lymphoma (NMZL), Extranodal Marginal Zone Lymphoma (ENMZL) of Mucosa-Associated Lymphoid Tissue (MALT), Splenic Marginal Zone Lymphoma (SMZL), LymphoPlasmacytic Lymphoma (LPL) and Small Lymphocytic Lymphoma (SLL). Follicular Lymphoma is the most indolent form and second most common form of all NHLs and they are a heterogeneous group of lymphoproliferative malignancies. Approximately 20% of all NHLs are Follicular Lymphomas. Advanced stage indolent NHL is not curable and as such, prolonging Progression Free Survival (PFS) and Overall Survival (OS), while maintaining Quality of Life, have been the goals of treatment intervention. Asymptomatic patients with indolent NHL are generally considered candidates for “watch and wait” approach. Patients with advanced stage symptomatic Follicular Lymphoma are often treated with induction chemoimmunotherapy followed by maintenance RITUXAN® (Rituximab). This can result in a median Progression Free Survival (PFS) of 6-8 yrs and a median Overall Survival of 12-15 yrs. However, approximately 30% of the patients will relapse in 3 years.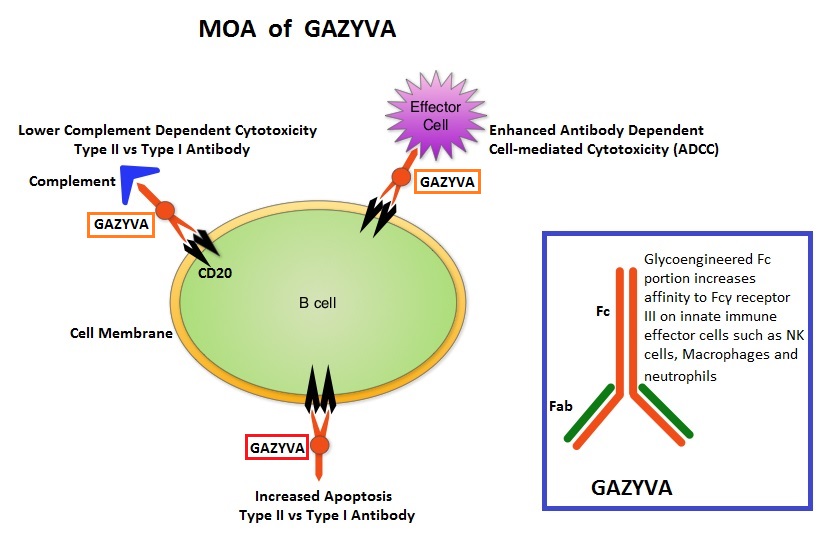
GAZYVA® (Obinutuzumab) is glycoengineered, fully humanized, third generation, type II anti-CD20 antibody (IgG1 monoclonal antibody) that selectivity binds to the extracellular domain of the CD20 antigen on malignant human B cells. By virtue of binding affinity of the glycoengineered Fc portion of GAZYVA® to Fcγ receptor III on innate immune effector cells (natural killer cells, macrophages and neutrophils), Antibody-Dependent Cell-mediated Cytotoxicity (ADCC) and Antibody-Dependent Cellular phagocytosis are significantly enhanced, but induces very little Complement-Dependent Cytotoxicity. This is in contrast to RITUXAN® which is a first generation type I, chimeric, anti-CD20 targeted monoclonal antibody that kills lymphoma cells primarily by Complement-Dependent Cytotoxicity and also ADCC.
GAZYVA® along with Bendamustine in the phase III GADOLIN study prolonged PFS, compared with Bendamustine alone, in patients with relapsed/refractory indolent Non Hodgkin lymphoma. Based on this promising data, the GALLIUM phase III trial was conducted in treatment naïve patients with Follicular Lymphoma. This study included 1,202 patients with newly diagnosed Follicular Lymphoma, who had Grade I-IIIa tumors and had an ECOG PS of 2 or less. Patients were randomized to receive either GAZYVA® plus chemotherapy, followed by GAZYVA® maintenance (N=601), or RITUXAN® plus chemotherapy, followed by RITUXAN® maintenance (N=601). The chemotherapy regimens used were CHOP, CVP or Bendamustine, based on the discretion of the treating physician. Patients received either RITUXAN® 375mg/m2 IV on day 1 of each cycle or GAZYVA® 1000 mg IV on days 1, 8, and 15 of cycle 1 and day 1 of subsequent cycles, for either eight 21-day cycles (CHOP and CVP) or six 28-day cycles (Bendamustine). Patients who achieved a Complete Response (CR) or Partial Response (PR) at the end of induction therapy, received maintenance therapy with RITUXAN® or GAZYVA® every 2 months for 2 years or until disease progression. The median age was 59 years and 57.1% of patients received Bendamustine, 33.1% received CHOP, and 9.8% received CVP. The primary endpoint was Progression Free Survival (PFS) and secondary endpoints included Response Rate, Overall Survival (OS), Disease Free Survival and safety. After a median follow up of 34.5 months, upon recommendations from the Independent Monitoring Committee, the study was unblinded after a preplanned interim efficacy analysis.
The estimated 3-year rate of Progression Free Survival in the GAZYVA® group was 80% compared with 73.3% in the RITUXAN® group, with a 34% reduction in the risk of progression or death noted in the GAZYVA® group (HR=0.66; P=0.001). There was however no difference between the two treatment groups in the 3-year Overall Survival (OS) rate (P=0.21). There was also no difference in the Response Rates between the two treatment groups ((88.5% in the GAZYVA® group and 86.9% in the RITUXAN® group). Patients treated with GAZYVA® had more serious adverse events, 46.1% versus 39.9% in the RITUXAN® group, but the discontinuation rate was similar in both treatment groups.
The authors concluded that for treatment naïve Follicular Lymphoma patients, combining GAZYVA® with chemotherapy resulted in a clinically meaningful improvement in PFS compared with RITUXAN® plus chemotherapy. Whether the improved Progression Free Survival in the GAZYVA® group is related to the maintenance treatment, remains to be explored. Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. Marcus R, Davies A, Ando K, et al. N Engl J Med 2017; 377:1331-1344
Targeting AKT Improves Outcomes in a Subset of Triple Negative Breast Cancer Patients
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. It is estimated that 252,710 new cases of invasive breast cancer and 63,410 new cases of non-invasive breast cancer will be diagnosed in women in 2017 and 40,610 women are expected to die from the disease. Triple Negative Breast Cancer (TNBC) is a heterogeneous, molecularly diverse group of breast cancers and are ER (Estrogen Receptor), PR (Progesterone Receptor) and HER2 (Human Epidermal Growth Factor Receptor 2) negative. TNBC accounts for 15% to 20% of invasive breast cancers, with a higher incidence noted in young patients. It is usually aggressive, and tumors tend to be high grade and patients with TNBC are at a higher risk of both local and distant recurrence. Those with metastatic disease have one of the worst prognoses of all cancers with a median Overall Survival of 13 months. The majority of patients with TNBC who develop metastatic disease do so within the first 3 years after diagnosis, whereas those without recurrence during this period of time have survival rates similar to those with ER-positive breast cancers. The lack of known recurrent oncogenic drivers in patients with metastatic TNBC, presents a major therapeutic challenge. Nonetheless, patients with TNBC often receive chemotherapy in the neoadjuvant, adjuvant or metastatic settings and approximately 30-40% of patients achieve a pathological Complete Response (pCR) in the neoadjuvant setting. Those who do not achieve a pathological Complete Response tend to have a poor prognosis. It therefore appears that there are subsets of patients with TNBC who may be inherently insensitive to cytotoxic chemotherapy. Three treatment approaches appear to be promising and they include immune therapies, PARP inhibition and inhibition of PI3K pathway.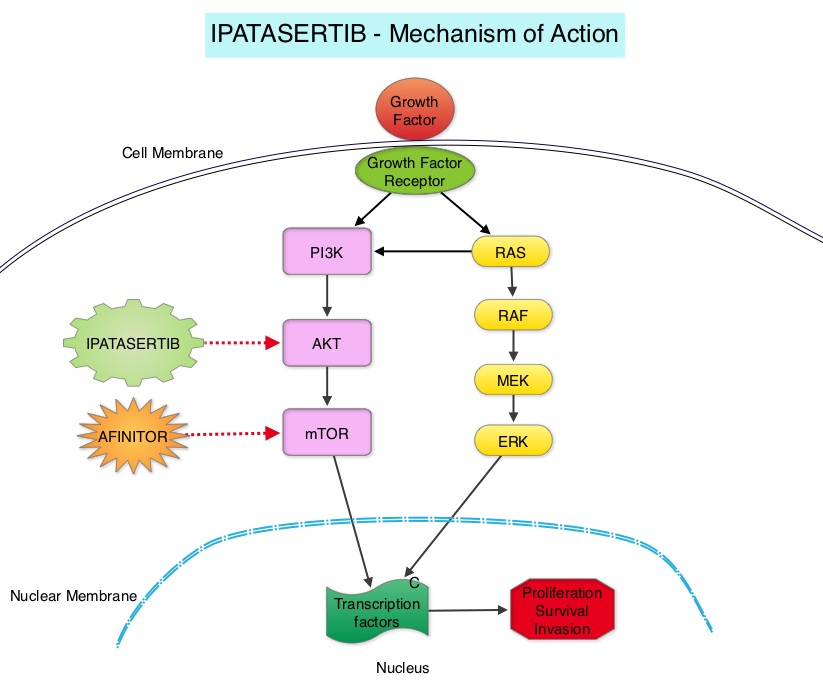
Using gene expression profiling, TNBC can be classified into 4 distinct molecular subtypes- two Basal-Like (BL1, BL2), Mesenchymal type (M) and Luminal Androgen Receptor type (LAR). BL1 molecular subtype of TNBCs are characterized by high levels of expression of genes involved in the cell cycle and DNA-damage repair pathways and accounts for up to 18% of TNBCs. These tumors are more sensitive to therapies targeting the DNA-repair pathways such as platinum based chemotherapy and Poly-ADP Ribose Polymerase (PARP) inhibition. BL2 molecular subtype of TNBCs represent 13% of TNBCs and in contrast are characterized by upregulation of growth factor signaling pathways, including the Epidermal Growth Factor (EGF), MET pathways, as well as genes involved in glycolysis and gluconeogenesis. These tumors may better respond to small molecule inhibitors of growth factor pathways. An alternate classification of Basal-Like subtype includes Basal-Like Immune Suppressed (BLIS) which is associated with downregulation of B cell, T cell, and Natural Killer cell immune-regulating pathways, and has the worse prognosis and Basal-Like Immune Activated (BLIA) subtype, which has the best prognosis due to upregulated immune-associated pathways. Mesenchymal TNBCs constitute approximately 10-30% of TNBC tumors and are associated with aberrations in the PI3K/AKT/ mTOR pathway as well as increased angiogenesis and may benefit from agents targeting these pathways. Metaplastic breast cancer belongs to this TNBC group. The Luminal Androgen Receptor (LAR) subtype accounts for approximately 11% of TNBCs. These tumors have a high expression of Androgen Receptor by IHC (Immuno HistoChemistry) and benefit from Androgen Receptor blockade and do not respond to cytotoxic chemotherapy.
Ipatasertib is a highly selective oral ATP-competitive, small-molecule, AKT inhibitor and sensitivity to Ipatasertib has been associated with high levels of phosphorylated AKT, PTEN protein loss or genetic mutations in PTEN, and PIK3CA mutations. KRAS and BRAF mutations are typically associated with resistance to Ipatasertib. It is estimated that approximately 50% of TNBCs have deficient expression of the tumor suppressor PTEN, which is associated with a higher degree of AKT pathway activation Preclinical studies showed synergy between Ipatasertib and Taxanes. Because of the high prevalence of PI3K/AKT pathway activation in TNBCs, the authors in this study evaluated the benefit of a combination of Ipatasertib and Paclitaxel as first line therapy, for TNBC.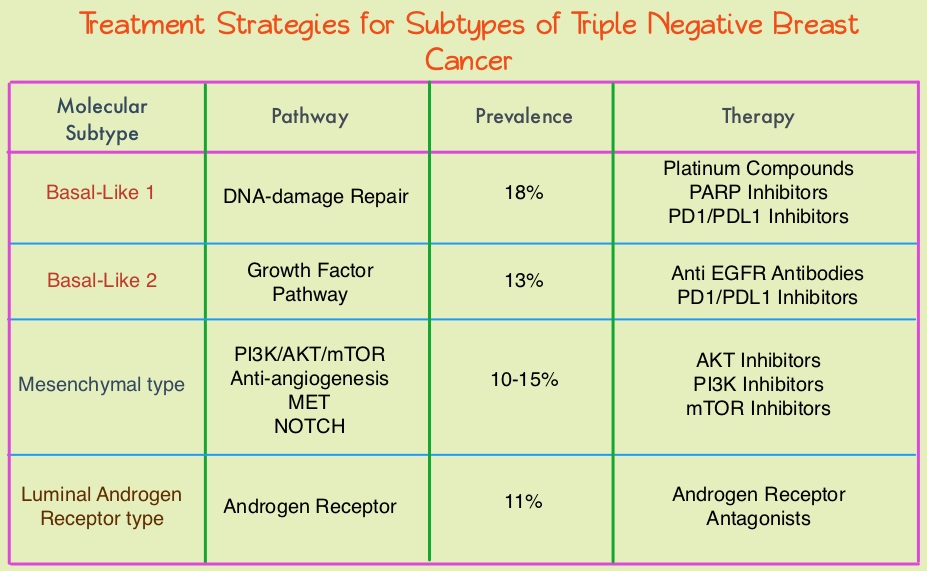
The LOTUS trial is a randomized, placebo controlled, double blind, phase II study in which 124 treatment naïve patients with inoperable, locally advanced or metastatic Triple Negative Breast Cancer were randomly assigned (1:1) to receive Paclitaxel 80 mg/m2 IV Days 1, 8, 15 of a 28 day cycle in combination with either Ipatasertib 400 mg PO daily (N=62) or placebo (N=62), administered on days 1-21 of each 28 day cycle. Treatment was continued until disease progression or unacceptable toxicity. Patients in this study were stratified based on expression of the PTEN tumor suppressor gene and alteration of PIK3CA/AKT1/PTEN in their tumors. The co-primary endpoints were Progression Free Survival (PFS) in the intent-to-treat population and Progression Free Survival in the PTEN-low population. Secondary endpoints included Objective Response Rate and Duration of Response. The median follow up was 10.3 months.
It was noted that the median PFS in the intent-to-treat population was 6.2 months with Ipatasertib versus 4.9 months with placebo (HR=0.60; P=0.037). In the 48 patients with low PTEN expression tumors, the median PFS however was 6.2 months with Ipatasertib and 3.7 months with placebo (HR=0.59; P=0.18), and this was not statistically significant. The PFS benefit was more pronounced in the patient group with PIK3CA/AKT1/PTEN-altered tumors, with a median PFS of 9.0 months in the Ipatasertib group versus 4.9 months in the placebo group (HR=0.44; P=0.041). The most common grade 3/4 adverse events in the Ipatasertib group were diarrhea and neutropenia.
The authors concluded that Ipatasertib prolonged Progression Free Survival compared to placebo and is the first study supporting AKT-targeted therapy for Triple Negative Breast Cancer, supporting the use of gene expression profiling in this heterogeneous malignancy. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Kim S, Dent R, Im S, et al on behalf of the LOTUS investigators. The Lancet Oncology 2017;18:1360-1372
African American Women with Benign Breast Disease and Triple Negative Breast Cancer
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. It is estimated that 252,710 new cases of invasive breast cancer and 63,410 new cases of non-invasive breast cancer will be diagnosed in women in 2017 and 40,610 women are expected to die from the disease. Benign Breast Disease (BBD) is an important risk factor for the later development of breast cancer in either breast. With the routine use of mammography, the identification of BBD has become more common, and this has in turn enabled the calculation of breast cancer risk. The Breast Cancer Risk Assessment Tool which is an interactive tool designed by researchers at the National Cancer Institute (NCI) and the National Surgical Adjuvant Breast and Bowel Project (NSABP) to estimate a woman's risk of developing invasive breast cancer, utilizes the number of breast biopsies and atypical hyperplasia, in addition to other factors, to estimate women’s risk of developing breast cancer. Benign Breast Disease (BBD) encompasses a variety of histologic entities. FibroCystic Changes (FCCs) constitute the most frequent benign disorder of the breast. FCCs may be multifocal and bilateral and women usually present with symptoms of breast pain and tender nodularities in breasts. Hormonal imbalance, with estrogen predominance over progesterone, has been implicated as an important contributing factor in its development. Fibrocystic changes are usually subdivided into nonproliferative lesions, proliferative lesions without atypia, and proliferative lesions with atypia (atypical hyperplasia). Proliferative or atypical lesions are associated with an increased risk of breast cancer. However a significant majority of breast biopsies (about 70%) show nonproliferative lesions.
Breast cancer is heterogeneous malignancy and using global gene expression analyses, 6 breast cancer intrinsic subtypes have been established. They include Luminal A, Luminal B, HER2-enriched, Claudin-low, Basal-like, and a Normal breast-like group. Triple Negative Breast Cancer (TNBC) accounts for 12–24% of all breast carcinomas and is negative for Estrogen Receptor (ER), Progesterone Receptor (PR), and HER2. There is a 2 fold increase in the incidence of TNBC among African American (AA) women compared with White American (WA) women and is a surrogate for the inherently aggressive Basal-like breast cancer subtype. This group has the worse prognosis compared to other breast cancer subtypes. Basal-like breast cancer sub-type is also a marker of hereditary breast cancer susceptibility. Multiparity may increase the risk of TNBC and reduce the likelihood of developing ER-positive breast cancer. Triple Negative Breast Cancer (TNBC) compared with non-TNBC, likely arises from different pathogenetic pathways. NF-κB pathway, which controls immune response, angiogenesis, the cell cycle, extracellular matrix degradation, and apoptosis, may represent a key regulator of TNBC. Activation of PI3K/AKT pathway in turn, has been implicated in oncogenic transformation.
Benign breast disease (BBD) has been associated with increased risk for ER positive/non-TNBC. The purpose of this study was to determine whether African American (AA) identity is associated with TNBC among a cohort of both AA and White American (WA) women, who were initially diagnosed with BBD. The authors in this study conducted a retrospective analysis of a cohort comprising 2588 African American women and 3566 White American women aged between 40 and 70 years, with a biopsy-proven Benign Breast Disease. This data was obtained from the Pathology Information System of Henry Ford Health System (HFHS), an integrated multihospital and multispecialty health care system in Detroit, Michigan. These individuals had breast biopsies performed between January 1, 1994, and December 31, 2005 and data analysis was performed from November 1, 2015, to June 15, 2016. Patients with prior breast cancer and those with breast cancer diagnosed within 6 months of BBD biopsy were excluded. Benign Breast Disease was classified as fibrocystic/proliferative/hyperplasia without atypia, with atypia, or with lobular carcinoma in situ. The mean follow up was 10.2 years for both AA patients and WA patients.
It was noted that more than 75% of the subsequent breast cancers in each subset were Ductal Carcinoma in Situ (DCIS) or Stage I. Among the African American (AA) patients who developed subsequent invasive breast cancer, 24.2% developed TNBC compared with 7.4% of the White American (WA) patients who developed subsequent invasive breast cancers (P=0.01). The 10 year probability estimate for developing TNBC was 0.56% for AA patients and 0.25% for WA patients.
The authors concluded that in this largest analysis to date of TNBC and its relationship to racial/ethnic identity and Benign Breast Disease as risk factors, African American identity persisted as a significant risk factor for Triple Negative Breast Cancer, suggesting that African American identity is associated with inherent susceptibility for TNBC pathogenetic pathways. This important finding should be taken into consideration, when discussing chemoprevention strategy among African American women with Benign Breast Diseases. Association Between Benign Breast Disease in African American and White American Women and Subsequent Triple-Negative Breast Cancer. Newman LA, Stark A, Chitale D, et al. JAMA Oncol. 2017;3:1102-1106.
FDA Approves OPDIVO® for Advanced Hepatocellular Carcinoma
SUMMARY: The FDA on September 22, 2017, granted accelerated approval to OPDIVO® (Nivolumab) for the treatment of HepatoCellular Carcinoma (HCC), in patients who have been previously treated with NEXAVAR® (Sorafenib). The American Cancer Society estimates that about 41,000 new cases of primary liver cancer will be diagnosed in the US for 2017 and 29,000 patients will die of their disease. Liver cancer is seen more often in men than in women and the incidence has more than tripled since1980. This increase has been attributed to the higher rate of hepatitis C virus (HCV) infection among baby boomers (born between 1945 through 1965). Obesity and type II diabetes have also likely contributed to the trend. Other risk factors include alcohol, which increases liver cancer risk by about 10% per drink per day, and tobacco use, which increases liver cancer risk by approximately 50%. HCC is the second most common cause of cancer-related deaths worldwide. NEXAVAR® (Sorafenib) was approved by the FDA in 2007 for the treatment of unresectable, HepatoCellular Carcinoma (HCC). Patients with advanced HCC who progress on NEXAVAR®, have a poor prognosis with limited treatment options.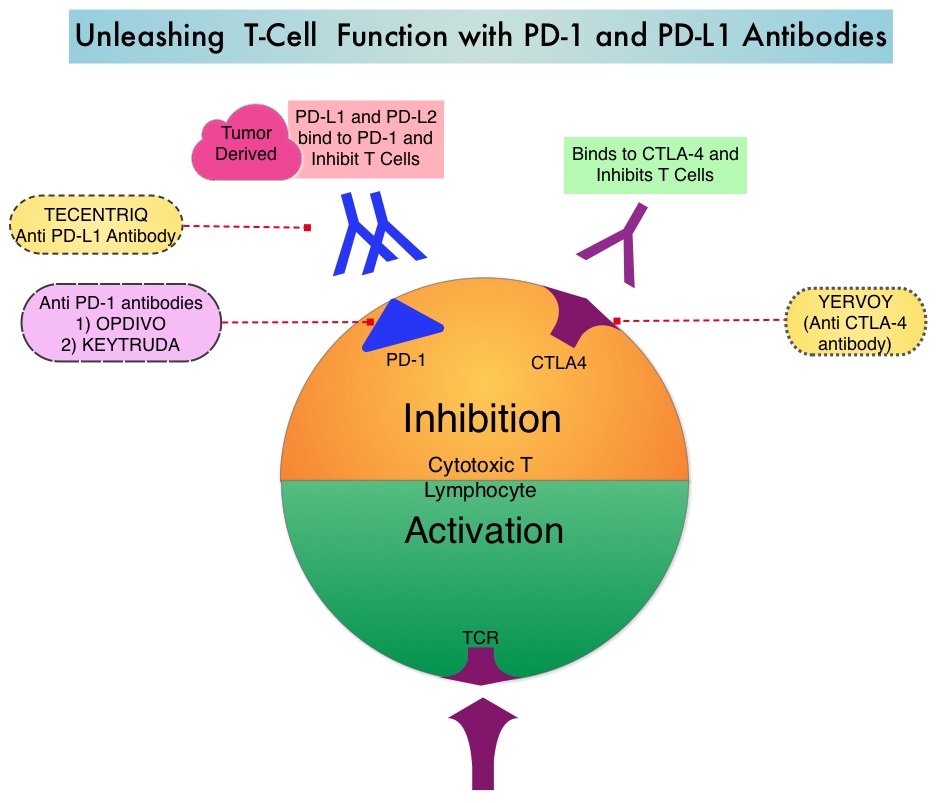
Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. Under normal circumstances, Immune checkpoints or gate keepers inhibit intense immune responses by switching off the T cells of the immune system and can thereby suppress antitumor immunity. These Immune checkpoint proteins/receptors include CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152), PD-1(Programmed cell Death 1), etc. Immune check point inhibitors are antibodies that target the immune checkpoint receptors and unleash the T cells, resulting in T cell proliferation, activation and a therapeutic response. OPDIVO® (Nivolumab) is an immune checkpoint PD-1 (Programmed cell Death 1) targeted, fully human, immunoglobulin G4 monoclonal antibody that demonstrated significant activity in HepatoCellular Carcinoma (HCC).
The approval of OPDIVO® in HCC was based on a 154-patient subgroup of CHECKMATE-040 study, which is a multicenter, open-label trial, conducted in patients with HCC and Child-Pugh A cirrhosis, who had progressed on or were intolerant to NEXAVAR®. Enrolled patients included patients without active viral hepatitis, patients with either active HBV-Hepatitis B Virus (31%) or HCV- Hepatitis C Virus (21%). Patients with active co-infection with HBV and HCV or with Hepatitis D Virus infection were excluded. Patients received OPDIVO® 3 mg/kg by IV infusion every 2 weeks. The median age was 64 years and 80% of patients were male. The primary endpoints were safety/tolerability and Objective Response Rate (ORR) by investigator and Blinded Independent Central Review. Secondary endpoints included Duration of Response, Disease Control Rate and Overall Survival.
The confirmed Objective Response Rate as assessed by Independent Central Review was 19 % with 3 Complete Responses and 19 partial responses(13%) and 41% of the patients had stable disease. Response duration ranged from 3 months to over 38 months and 91% of responders had responses lasting 6 months or longer and 55% of the patients had responses lasting 12 months or longer. Overall responses were noted regardless of tumor cell PD-L1 expression and chronic hepatitis virus infection. Common adverse reactions included fever, rash, musculoskeletal pain, nausea, diarrhea, cough and dyspnea. There was a higher incidence of elevations in transaminases and bilirubin levels noted.
It was concluded that OPDIVO® demonstrated durable responses with long-term survival and favorable safety in patients with advanced HCC. This FDA approval provides a new treatment option for appropriate patients with HCC, who progress on prior systemic therapy. STIVARGA® (Regorafenib) is another agent presently available for this subset of patients. Nivolumab (nivo) in sorafenib (sor)-naive and -experienced pts with advanced hepatocellular carcinoma (HCC): CheckMate 040 study. Crocenzi TS, El-Khoueiry AB, Yau TC, et al. J Clin Oncol. 2017;35 (suppl; abstr 4013).
FDA Approves TAFINLAR® and MEKINIST® Combo for BRAF V600E-Mutant Non Small Cell Lung Cancer
SUMMARY: The FDA on June 22, 2017 granted regular approvals to TAFINLAR® ((Dabrafenib) and MEKINIST® (Trametinib) administered in combination, for patients with metastatic Non Small Cell Lung Cancer (NSCLC) with BRAF V600E mutation, as detected by an FDA-approved test. These are the first FDA approvals specifically for treatment of patients with BRAF V600E mutation-positive metastatic NSCLC. The FDA also approved the Oncomine® Dx Target Test (Thermo Fisher Scientific), a Next Generation Sequencing (NGS) test to detect multiple gene mutations for lung cancer in a single test from a single tissue specimen. This test detects the presence of BRAF, ROS1, and EGFR gene mutations or alterations in tumor tissue of patients with NSCLC. This test can be used to select patients with NSCLC with the BRAF V600E mutation for treatment with the combination of TAFINLAR® and MEKINIST®. This is the first NGS oncology panel test approved by the FDA for multiple companion diagnostic indications. Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2017 about 222,500 new cases of lung cancer will be diagnosed and over 155,000 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States.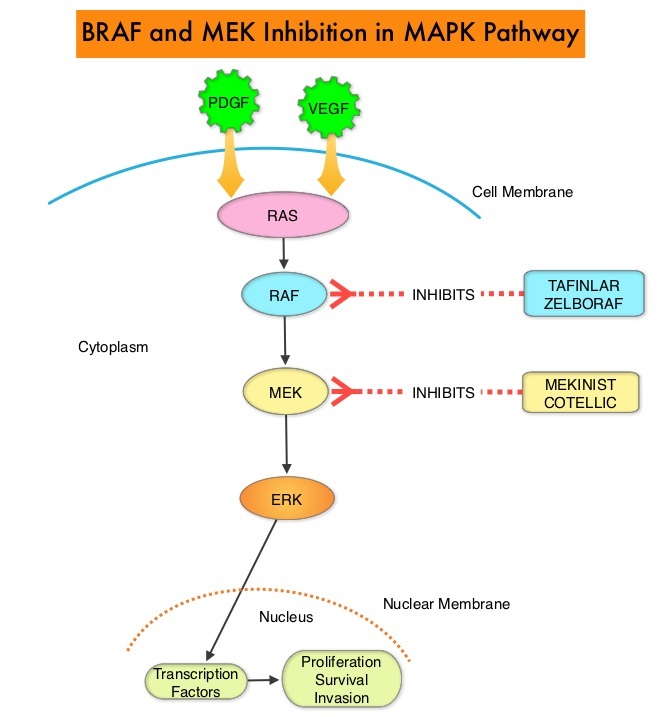
The approval of the combination of MEKINIST® (Trametinib) and TAFINLAR® (Dabrafenib), to treat patients with advanced NSCLC, was based on the understanding of the biological pathways of this malignancy. The Mitogen-Activated Protein Kinase pathway (MAPK pathway) is an important signaling pathway which enables the cell to respond to external stimuli. This pathway plays a dual role, regulating cytokine production and participating in cytokine dependent signaling cascade. The MAPK pathway of interest is the RAS-RAF-MEK-ERK pathway. The RAF family of kinases includes ARAF, BRAF and CRAF signaling molecules. BRAF is a very important intermediary of the RAS-RAF-MEK-ERK pathway. BRAF mutations have been demonstrated in 6%-8% of all malignancies and BRAF V600E mutation occurs in 1-2% of lung adenocarcinomas and acts as an oncogenic driver.
BRF113928 is an open-label, multicohort, multicentre, non-randomized, phase II study which sequentially enrolled patients with BRAF V600E mutation-positive metastatic NSCLC, across 3 cohorts. The first 2 cohorts included previously treated patients. The median age was 64 years, 98% had adenocarcinoma, and majority of patients (72%) were former or current smokers. In the first cohort, 84 patients received single agent TAFINLAR® following one or more prior platinum-based chemotherapy. In the second cohort, 57 patients received a combination of TAFINLAR® 150 mg orally twice daily and MEKINIST® 2 mg orally once daily. Patients in this second cohort had received at least 1 prior line of platinum-based chemotherapy regimen with disease progression and 33% had received 2 or more prior chemotherapy regimens.
It was noted that in these previously treated cohorts of patients, the Objective Response Rate (ORR) for the combination treatment based on independent review was 63%, with a median Duration of Response (DoR) of 12.6 months. The ORR for patients who received single agent TAFINLAR® was 27% and the median DoR was 9.9 months. The investigator assessed median Progression Free Survival (PFS) was 10.2 months, and median Overall Survival (OS) was 18.2 months.
The authors in this latest publication reported the the efficacy and safety of TAFINLAR® plus MEKINIST® treatment in previously untreated patients with BRAF V600E-mutant metastatic NSCLC (third cohort). This cohort enrolled 36 patients and patients received TAFINLAR® 150 mg orally twice daily plus MEKINIST® 2 mg orally once daily, until disease progression or unacceptable toxicities. The median follow up was 15.9 months. The Primary endpoint was investigator assessed ORR. Secondary endpoints included Duration of Response (DoR), PFS, OS, and safety.
The Objective Response Rate in this cohort was 64%, with 6% Complete Response and 58% Partial Response. The Disease Control Rate was 75%. The median Duration of Response was 10.4 months, median PFS was 10.9 months and median OS was 24.6 months. The most common grade 3 or 4 adverse events were pyrexia, liver function abnormalities, hypertension and vomiting.
The authors concluded that TAFINLAR® plus MEKINIST® demonstrated substantial antitumor activity and durable responses in patients with treatment-naive BRAF V600E-mutant NSCLC. This study confirmed that there is a fourth actionable biomarker, BRAF V600E, in addition to EGFR, ALK and ROS-1, for patients with Non Small Cell Lung Cancer. Dabrafenib plus trametinib in patients with previously untreated BRAF V600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Planchard D, Smit EF, Groen HJ, et al. The Lancet Oncology 2017;18:1307-1316
FDA Approves VERZENIO® for Hormone Receptor Positive, HER2-Negative Breast Cancer
SUMMARY: The FDA on September 28, 2017, approved VERZENIO® (Abemaciclib) in combination with FASLODEX® (Fulvestrant) for women with Hormone Receptor positive (HR-positive), HER2-negative, advanced or metastatic breast cancer, with disease progression following endocrine therapy. In addition, VERZENIO® was approved as monotherapy for women and men with HR-positive, HER2-negative advanced or metastatic breast cancer with disease progression following endocrine therapy and prior chemotherapy in the metastatic setting. Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. It is estimated that 252,710 new cases of invasive breast cancer and 63,410 new cases of non-invasive breast cancer will be diagnosed in women in 2017 and 40,610 women are expected to die from the disease.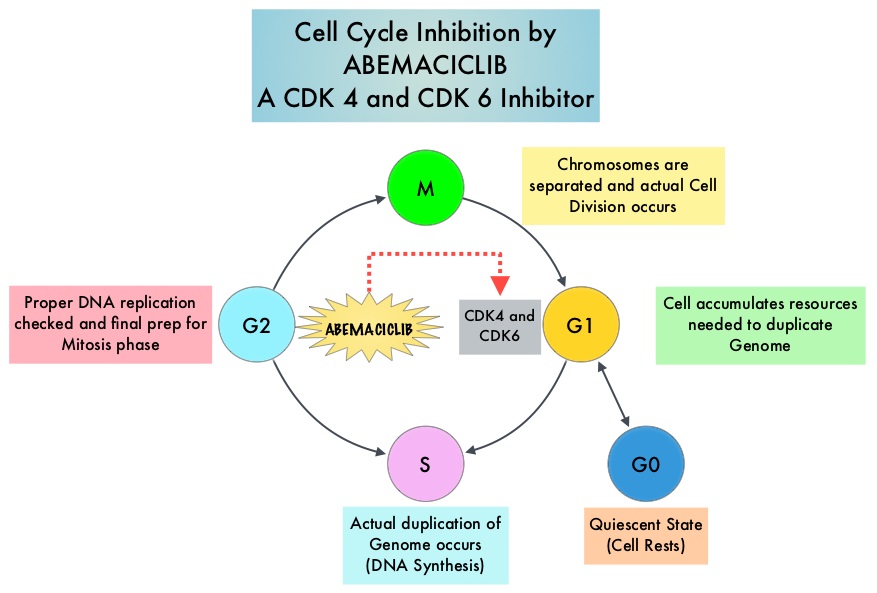
Cyclin Dependent Kinases (CDK) play a very important role to facilitate orderly and controlled progression of the cell cycle. Genetic alterations in these kinases and their regulatory proteins have been implicated in various malignancies. Cyclin Dependent Kinases 4 and 6 (CDK4 and CDK6), phosphorylate RetinoBlastoma protein (RB), and initiate transition from the G1 phase to the S phase of the cell cycle. RetinoBlastoma protein has antiproliferative and tumor-suppressor activity and phosphorylation of RB protein nullifies its beneficial activities. CDK4 and CDK6 are activated in hormone receptor positive breast cancer, promoting breast cancer cell proliferation. Further, there is evidence to suggest that endocrine resistant breast cancer cell lines depend on CDK4 for cell proliferation. The understanding of the role of Cyclin Dependent Kinases in the cell cycle, has paved the way for the development of CDK inhibitors.
VERZENIO® is an oral, selective inhibitor of CDK4 and CDK6 kinase activity, and prevents the phosphorylation and subsequent inactivation of the Rb tumor suppressor protein, thereby inducing G1 cell cycle arrest and inhibition of cell proliferation. VERZENIO® is structurally distinct from other CDK 4 and 6 inhibitors (such as Ribociclib and Palbociclib) and is 14 times more potent against cyclin D1/CDK 4 and cyclin D3/CDK 6, in enzymatic assays. VERZENIO® in the phase I trials was noted to be active in HR-positive, metastatic breast cancer, as monotherapy as well as in combination with FASLODEX®. Based on this preliminary data, two clinical trials, MONARCH 1 and MONARCH 2, were conducted.
The approval of VERZENIO® as monotherapy in HR-positive, metastatic breast cancer was based on MONARCH 1, which is a a single-arm, open-label, phase II, multicenter study, which enrolled women with measurable HR-positive, HER2-negative metastatic breast cancer, whose disease progressed during or after endocrine therapy, had received a taxane in any setting, and who received one or two prior chemotherapy regimens in the metastatic setting. This trial included 132 patients who received VERZENIO® 200 mg orally twice daily on a continuous schedule, until disease progression. The Objective Response Rate was 19.7%, with a median response duration of 8.6 months.
The approval of VERZENIO® in combination with FASLODEX® was based on MONARCH 2, which is an international, double-blind, phase III study in which 669 patients were randomized in a 2:1 ratio to receive either VERZENIO® plus FASLODEX® (N=446) or placebo plus FASLODEX® (N=223). Enrolled patients had HR-positive, HER2-negative metastatic breast cancer, with disease progression while receiving neoadjuvant or adjuvant endocrine therapy, within 12 months of adjuvant endocrine therapy, or while receiving endocrine therapy for metastatic breast cancer. Patients must not have received more than one endocrine therapy or any prior chemotherapy for metastatic breast cancer. Randomized patients received either VERZENIO® 150 mg or placebo orally twice daily plus FASLODEX® 500 mg IM on Day 1 and Day 15 of cycle 1 and then on Day 1 of cycle 2 and beyond (28-day cycles). Treatment was continued until disease progression or unmanageable toxicities. The mean patient age was 60 years, 82% of patients were postmenopausal, 72% had measurable disease, 56% had visceral disease, and 25% had primary endocrine therapy resistance. About 60% of patients had received chemotherapy in the adjuvant or neoadjuvant setting and 69% of the patients had prior therapy with Aromatase Inhibitors (AI). The Primary end point was Progression Free Survival (PFS), and Secondary end points included Overall Survival (OS), Objective Response Rate (ORR), Duration of Response, Clinical Benefit Rate, Quality of Life, and safety.
The median PFS for the group receiving VERZENIO® plus FASLODEX® was 16.4 months compared with 9.3 months for those taking placebo with FASLODEX® (HR= 0.55; P<0.0001). In patients with measurable disease, the Objective Response Rate for the group receiving VERZENIO® plus FASLODEX® was 48.1% compared to 21.3% in the placebo with FASLODEX® treated patients. The most common adverse events in the VERZENIO® versus placebo groups were diarrhea neutropenia, nausea and fatigue.
It was concluded that a combination of VERZENIO® plus FASLODEX® significantly improved Progression Free Survival and Objective Response Rates, with a tolerable safety profile, in patients with Hormone Receptor-positive and HER 2-negative metastatic breast cancer who progressed while receiving endocrine therapy. MONARCH 2: Abemaciclib in Combination With Fulvestrant in Women With HR+/HER2− Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. Sledge GW, Toi M, Neven P, et al. DOI: 10.1200/JCO.2017.73.7585 Journal of Clinical Oncology 35, no. 25 (September 2017) 2875-2884.
MONARCH 1: Results from a phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as monotherapy, in patients with HR+/HER2- breast cancer, after chemotherapy for advanced disease. Dickler MN, Tolaney SM, Rugo HS, et al. J Clin Oncol 34, 2016 (suppl; abstr 510).
VERZENIO® (Abemaciclib)
The FDA on September 28, 2017 approved VERZENIO® in combination with Fulvestrant for women with HR-positive, HER2-negative advanced or metastatic breast cancer, with disease progression following endocrine therapy. In addition, VERZENIO® was approved as monotherapy, for women and men with HR-positive, HER2-negative advanced or metastatic breast cancer, with disease progression following endocrine therapy and prior chemotherapy in the metastatic setting. VERZENIO® is a product of Eli Lilly and Company.