SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2017 about 222,500 new cases of lung cancer will be diagnosed and over 155,000 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Approximately one third of all patients with NSCLC have stage III, locally advanced disease at the time of initial presentation. Worldwide, about 500,000 patients are diagnosed with unresectable, stage III NSCLC, each year. These patients include those with locally advanced primary tumors with tumor invading the vital mediastinal organs, as well as those with involvement of locoregional mediastinal lymph nodes. These patients are often treated with platinum-based doublet chemotherapy with concurrent radiation and have a median Progression Free Survival (PFS) of approximately 8 months and 5 year survival of only 15%. There is hence a significant unmet need for this patient group, with no major treatment advances thus far.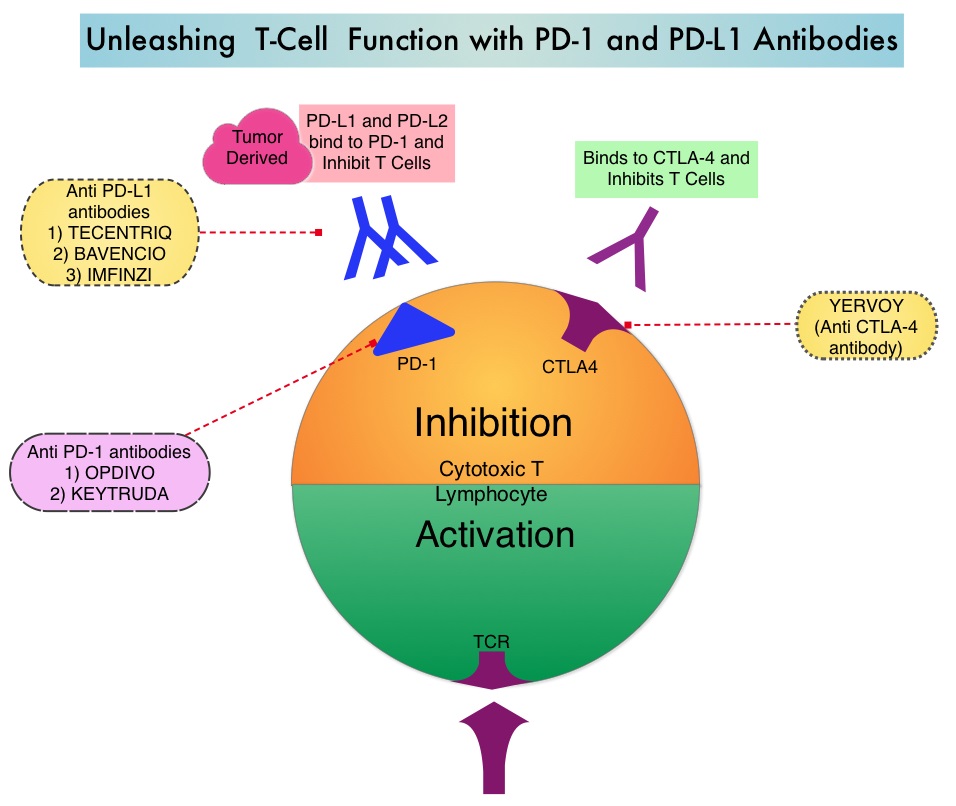
Preclinical evidence had suggested that chemotherapy and radiotherapy may upregulate PD-L1 expression in tumor cells. IMFINZI® (Durvalumab) is a selective, high-affinity, human IgG1 monoclonal antibody, that blocks the binding of Programmed Death Ligand 1 (PD-L1) to Programmed Death 1 (PD-1) and CD80, thereby unleashing the T cells to recognize and kill tumor cells. IMFINZI® showed encouraging antitumor activity in an early phase clinical study involving multiple advanced solid tumors, including stage IIIB or IV NSCLC. IMFINZI® was recently approved by the FDA for the treatment of patients with locally advanced or metastatic urothelial carcinoma, who had received prior platinum-based chemotherapy.
The authors in this publication evaluated the role of immune checkpoint blockade in locally advanced, unresectable, stage III NSCLC. PACIFIC trial is a randomized, double-blind, international, phase III study in which IMFINZI® as consolidation therapy was compared with placebo, in patients with stage III, locally advanced, unresectable NSCLC, that had not progressed following platinum-based chemoradiotherapy. Eligible patients received two or more cycles of platinum-based doublet chemotherapy concurrently with definitive radiation therapy (54-66 Gy). Following completion of concurrent chemoradiation treatment, 713 patients were randomized, of whom 709 patients in a 2:1 ratio received consolidation treatment, within 6 weeks after completion of chemoradiation with IMFINZI® 10 mg/kg every 2 weeks (N=473) or placebo (N=236), for up to 12 months. The median age was 64 years, and the majority of patients were men (70%) and 46% had a squamous histology. The coprimary end points were Progression Free Survival (PFS) and Overall Survival (OS). Secondary end points included 12-month and 18-month PFS rates, Objective Response Rate (ORR), Duration of Response, time to death or distant metastasis, and safety. The authors reported the results of a preplanned interim analysis after a median follow up of 14.5 months.
The median PFS from randomization to consolidation treatment was 16.8 months with IMFINZI® versus 5.6 months with placebo (HR=0.52; P<0.001). This meant a 48% decrease in the probability of disease progression with IMFINZI® and this improvement was consistent across all patient subgroups that were analyzed. The 12-month PFS was 55.9% vs 35.3%, and the 18-month PFS rate was 44.2% vs 27.0%, in favor of IMFINZI®. The ORR was higher with IMFINZI® compared to placebo (28.4% vs 16.0%; P<0.001), and the median Duration of Response was longer as well, with 73% of the patients in the IMFINZI® group having an ongoing response at 18 months versus 47% of the patients in the placebo group. Patients in the IMFINZI® group also had a lower incidence of new brain metastases. The median time to death or distant metastasis was longer with IMFINZI® compared with placebo (23.2 months vs 14.6 months; P<0.001). Adverse events of any grade occurred in 68% of patients in the IMFINZI® group compared to 53% in the placebo group and majority of the toxicities were grade 1 or 2, and grade 3 or higher toxicities were infrequent (less than10%), in both treatment groups. Treatment had to be discontinued due to pneumonitis in 6.3% of patients on IMFINZI® and 4.3% on placebo.
It was concluded that IMFINZI® significantly prolonged PFS in all prespecified groups of patients with locally advanced stage III NSCLC, and toxicity profile was acceptable. Biomarkers, such as mutational load or immunosignature, may be of value, as PD-L1 expression had little or no impact on outcomes. The National Comprehensive Cancer Network (NCCN) Guidelines have been updated to include one year of consolidation therapy with IMFINZI®, after curative-intent chemoradiation, for inoperable stage III lung cancer. Durvalumab after Chemoradiotherapy in Stage III Non-Small Cell Lung Cancer. Antonia SJ, Villegas A, Daniel D, et al. for the PACIFIC Investigators. N Engl J Med 2017; 377:1919-1929