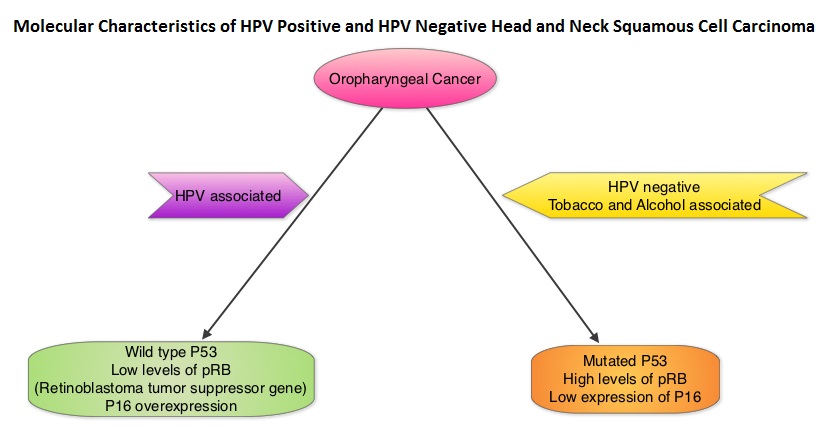
SUMMARY: Glioblastoma Multiforme (GBM)) is the most common malignant tumor of the central nervous system in adults and originates in astrocytes which are specialized glial cells. It is estimated that approximately 12,500-18,000 new cases of GBM are diagnosed annually in the United States. The most frequent location for GBM is cerebral hemispheres and 95% of these tumors arise in supratentorial region. The etiology remains unclear and genetic predisposition has been observed in only 5-10 % of cases. GBM can be primary when arising de novo without clinical and histological evidences of a precursor lesion or secondary when they progress slowly from preexisting lower-grade astrocytoma. Primary GBMs are associated with hallmark genetic alterations and they include Epidermal Growth Factor Receptor (EGFR) gene mutation and amplification, over expression of Mouse Double Minute 2 (MDM2), deletion of p16 and Loss of Heterozygosity (LOH) of chromosome 10q holding Phosphatase and Tensin homolog (PTEN) and TERT promoter mutation.
GBM is not curable and management includes maximal safe debulking of the tumor followed by concurrent chemoradiation and then adjuvant chemotherapy. Current chemotherapy agents with FDA approval for treatment of GBM include Temozolomide, Bevacizumab (AVASTIN®), Lomustine (GLEOSTINE®), Carmustine (BiCNU®) and Carmustine Implant (GLIADEL® Wafer for intraoperative implantation). The median survival for GBM patients from diagnosis, with the current therapies, is about 15 months.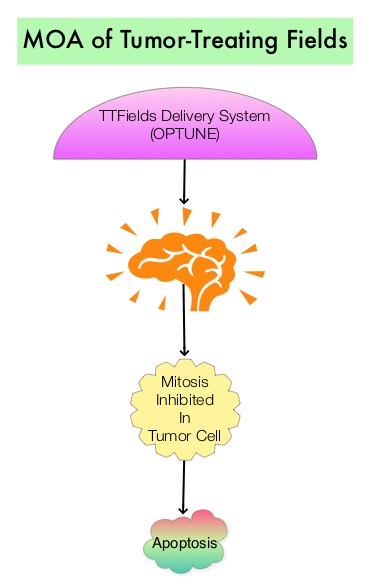
Tumor-Treating Fields (TTFields) delivery system (OPTUNE®) is a novel external therapeutic device that slows and reverses tumor growth by inhibiting mitosis. The battery operated-TTF delivery system generates low intensity, intermediate frequency, alternating electrical fields to the brain. These electrical fields exert selective toxicity in dividing cells by interfering with organelle assembly in the cell and thereby facilitates apoptosis (programmed cell death), by preventing cell division. The non-dividing cells are not affected by these electrical fields. Tumor Treating Fields (TTFields) delivery system, OPTUNE®, along with Temozolomide is presently approved by the FDA for the treatment of adult patients with newly diagnosed, supratentorial Glioblastoma (GBM) following maximal debulking surgery and radiation with concurrent chemotherapy. It is also approved for the treatment of recurrent GBM as a monotherapy, after surgical and radiation options have been exhausted. Insulated ceramic discs (transducer arrays) are placed directly on the scalp and held by adhesive bandages. They deliver the electrical fields after they are connected to the TTFields delivery system, OPTUNE®. Patients wear the device for at least 18 hours a day and for at least four weeks.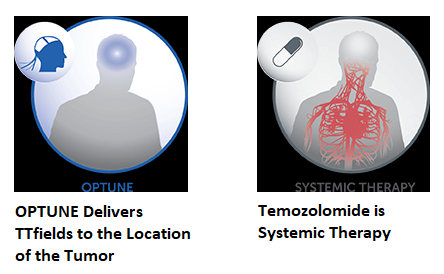
Previously published studies had shown that TTFields, in addition to its antimitotic effect on the dividing cell, can augment response to alkylator-based chemotherapy. These durable responses were sometimes delayed after an initial progression, and there was a high correlation between treatment compliance and survival. The EF-14 trial is a randomized, multicenter, open-label, phase III study, in which 695 patients with newly diagnosed grade IV GBM, who had completed standard treatment with surgery (resection or biopsy) and concurrent chemoradiation with Temozolomide, were randomized within 7 weeks of their last radiation dose, in a 2:1 ratio to TTFields plus maintenance Temozolomide chemotherapy (N=466) or Temozolomide alone (N=229). The TTFields, consisted of low-intensity, 200 kHz frequency, alternating electric fields and was delivered 18 hours/day or more, via 4 transducer arrays on the shaved scalp and connected to a portable device. Temozolomide was administered to both treatment groups at 150-200 mg/m2 PO for 5 days per 28 day cycle for 6-12 cycles. Patients had a Karnofsky Performance Score of 70 or more, with supratentorial tumor location and non-progressive disease. Patients were excluded if the tumor location was infratentorial and if there was evidence of increased intracranial pressure. The median age was 56 years, and majority of the patients were male. The Primary endpoint was Progression Free Survival (PFS) and the Secondary endpoint was Overall Survival (OS).
A preliminary report from this trial was published in 2015 and the authors now report the results of their final analysis. The median PFS from randomization was 6.7 months in the TTFields plus Temozolomide group and 4.0 months in the Temozolomide alone group (HR=0.63; P<0.001). The median OS was 20.9 months in the TTFields plus Temozolomide group versus 16.0 months in the Temozolomide alone group (HR=0.63; P< 0.001). This meant a 37% improvement in PFS and OS for patients who received TTFields plus Temozolomide compared to patients who received Temozolomide alone. The statistically significant benefit of TTFields with Temozolomide on Overall Survival, was seen in all pre-specified patient subgroups, regardless of prognostic factors such as age, performance status, MGMT promotor methylation and extent of resection. Mild to moderate skin toxicity underneath the transducer arrays occurred in 52% of patients in the TTFields plus Temozolomide group.
The authors concluded that the results in the final analysis were consistent with the previous interim analysis results, and for GBM patients who had received standard chemoradiation therapy, the addition of TTFields to maintenance Temozolomide chemotherapy, resulted in statistically significant improvement in PFS and OS, compared with maintenance Temozolomide alone. This is the first positive phase III trial in newly diagnosed GBM, since the efficacy of Temozolomide was established in 2005. Stupp R, Tailibert S, Kanner A, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA. 2017;318:2306-2316