Perioperative Interruption of Direct Oral Anticoagulants in Patients with Venous Thromboembolic Disease
SUMMARY: Direct Oral AntiCoagulants (DOACs) are often prescribed for thromboembolic events. This class of anticoagulants, have a rapid onset and offset of action, short half-life and predictable anticoagulant effects, without the need for routine monitoring. Further, several studies have demonstrated non-inferiority or superiority of this class of drugs compared with Vitamin K Aantagonists (VKAs), with regards to prevention and treatment of thromboembolic events. It is estimated that each year 10-15% of patients on DOACs will undergo an invasive procedure or surgery and will require temporary interruption of anticoagulation prior to standard-risk procedures and procedures with increased risk for bleeding. Several studies have evaluated the perioperative interruption of DOACs based on half-life of the anticoagulant and the underlying procedural bleeding risk in patient with Atrial Fibrillation. Whether these findings can be extrapolated to patients with VTE, has remained unclear.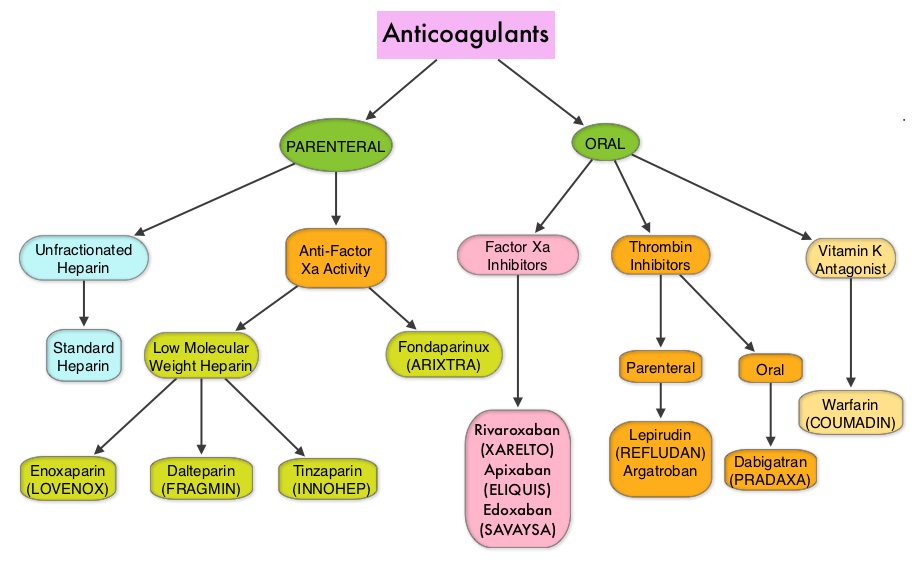
The authors in this study evaluated the thrombotic and bleeding outcomes following the perioperative interruption of Direct Oral AntiCoagulants, in patients with prior VTE (Venous ThromboEmbolism). This retrospective study included 190 patients who were on Direct Oral AntiCoagulants, such as PRADAXA® (Dabigatran), XARELTO® (Rivaroxaban), SAVAYSA® (Edoxaban) or ELIQUIS® (Apixaban), for previous VTE, and were scheduled to undergo an invasive procedure or surgery. They required temporary interruption of anticoagulation prior to standard-risk procedures and procedures with increased risk for bleeding. About 80% of the patients had unprovoked VTE, as the most recent thrombotic event, and 25% of the patients had recurrent VTE. The mean age was 59 years.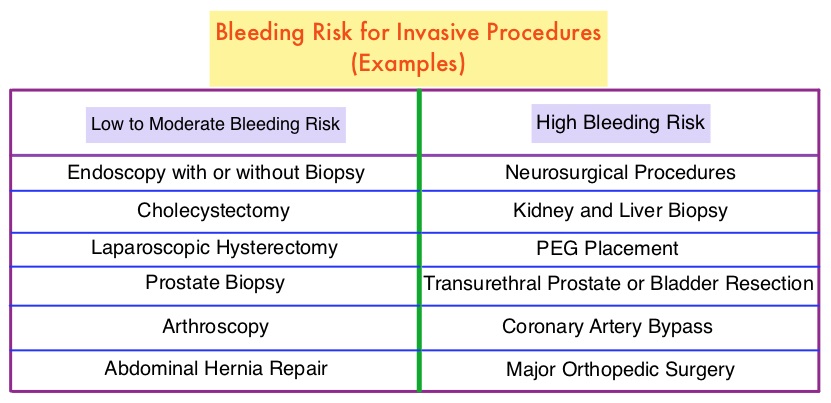
The timing to interrupt and reinitiate Direct Oral Anticoagulant therapy was at the discretion of the treating physician, and typically DOACs were held for three half-lives prior to and restarted 2 days following standard-bleeding risk procedures, and for five half-lives prior to and restarted 4 days following high-bleeding risk procedures. The mean time from last dose of DOAC to surgery was 56.9 hours for standard-bleeding risk procedures and 69.9 hours for high-bleeding risk procedures. The mean time to therapeutic dosing of DOACs was 47 hours in the standard-bleeding risk group and 80.2 hours in the high-bleeding risk group. Approximately 41% of patients also received prophylactic doses of Low Molecular Weight Heparin or DOACs in the immediate postoperative period before re-initiation of therapeutic anticoagulation. Also taken into consideration was the elimination half-life of DOACs which can be increased by decreased renal function (PRADAXA® > SAVAYSA® > XARELTO® and ELIQUIS®), severe hepatic insufficiency (XARELTO® and ELIQUIS® > SAVAYSA® > PRADAXA®) and drug interactions. The Primary efficacy outcome was the 30-day symptomatic VTE rate, and the Primary safety outcome was the 30-day major bleeding rate. Secondary outcomes included overall mortality and the rate of clinically relevant non-major bleeding.
The 30-day VTE rate was 1.05% and the 30-day major bleeding rate was 0.53%. There were no deaths during the 30-day follow-up period. The rate of clinically relevant non-major bleeding was 3.16%.
It was concluded that perioperative interruption of Direct Oral AntiCoagulants, using a strategy that considered the half-life of the DOAC and the underlying procedural bleeding risk, appeared to be both safe and effective among patients with prior Venous ThromboEmbolism. Thrombotic and bleeding outcomes following perioperative interruption of direct oral anticoagulants in patients with venous thromboembolic disease. Shaw J, de Wit C, Le Gal G, et al. J Thromb Haemost. 2017;doi:10.1111/jth.13670