
The FDA on April 19, 2018 approved TAGRISSO® for the first-line treatment of patients with metastatic Non Small Cell Lung Cancer (NSCLC), whose tumors have Epidermal Growth Factor Receptor (EGFR) exon 19 deletions or exon 21 L858R mutations, as detected by an FDA-approved test. TAGRISSO® is a product of AstraZeneca Pharmaceuticals LP.
Month: April 2018
TAVALISSE® (Fostamatinib disodium hexahydrate)
The FDA on April 17, 2018 approved TAVALISSE® for the treatment of thrombocytopenia in adult patients with chronic Immune Thrombocytopenia (ITP) who have had an insufficient response to a previous treatment. TAVALISSE® is a product of Rigel Pharmaceuticals, Inc.
OPDIVO® (Nivolumab) and YERVOY® (Ipilimumab)
The FDA on April 16, 2018 granted approvals to OPDIVO® and YERVOY® in combination, for the treatment of intermediate or poor risk, previously untreated advanced Renal Cell Carcinoma. OPDIVO® and YERVOY® are products of Bristol-Myers Squibb Co.
FDA Approves TAVALISSE® for Chronic ITP
SUMMARY: The FDA on April 17, 2018, approved TAVALISSE® (Fostamatinib tablets) for the treatment of thrombocytopenia in adult patients with chronic Immune Thrombocytopenic Purpura (ITP), who have had an insufficient response to a previous treatment. ITP can manifest as an acute self limited disease often seen in children (Acute ITP) or in a chronic form (Chronic ITP) seen in adults with the thrombocytopenia lasting for 6 months or longer.
TAVALISSE is an oral Tyrosine Kinase Inhibitor that targets SYK kinase (Spleen Tyrosine Kinase). SYK associates with Fcγ receptors (FcγR) on the cell surface of various inflammatory cells, including macrophages, which are in turn responsible for platelet clearance in ITP. Inhibition of SYK by the active metabolite of TAVALISSE® reduces the destruction of platelets by macrophages, that are activated in the immune system. TAVALISSE® is the first and only SYK inhibitor indicated for adults with Chronic ITP and the present study validated the therapeutic effect of SYK inhibition in an autoimmune disease.
The approval of TAVALISSE® was supported by data from two randomized placebo-controlled phase III trials and an open label extension study, as well as an initial proof-of-concept study. FIT-1 and FIT-2 are two identical, multicenter, randomized, double-blind, placebo-controlled, phase III trials that included 150 patients with persistent or Chronic ITP, who had an insufficient response to previous treatment, which included Corticosteroids, Immunoglobulins, Splenectomy, and/or a Thrombopoietin Receptor Agonist (TPO-RA). Patients were randomized 2:1 to receive either TAVALISSE® 100 mg orally twice daily or placebo for 24 weeks. Dose could be escalated to 150 mg orally twice daily after one month. The median age was 54 years and enrolled patients had three documented platelet counts of 30,000/µL or less. Patients had ITP for a median of 8.5 years prior to enrollment and the median baseline platelet count was 16,000/μL. The most common prior treatments for ITP were Steroids (94%), TPO-RAs (47%), Splenectomy (35%), and Rituximab (32%). The Primary endpoint was Stable platelet response of 50,000/ µL or more on at least 4 of the 6 biweekly visits between Weeks 14 and 24 of the study, without rescue treatment.
The Overall Response Rate with TAVALISSE® in the two trials, FIT-1 and FIT-2 was 29% versus 2% in the placebo group (P<0.001). Among the responders on TAVALISSE®, Stable Response (Primary end point) was noted in 18% versus 1% in the placebo group (P=0.007) and Intermediate Response (defined as at least 2 consecutive biweekly platelet counts of 50,000/μL or more, without rescue treatment) was noted in 11% versus 0% in the placebo group (P<0.001).
Among patients treated with TAVALISSE®, the median platelet counts at 24 weeks of follow up was 95,000/μL for those who had a Stable Response, 49,000/μL for those who had Intermediate Response, and 20,500/μL in non-responders. For those in the placebo group, however, median platelet counts only reached 17,500/μL. More than half of patients treated with TAVALISSE® (54%) had increment in platelet count (20,000/μL or more), compared with 29% of patients receiving placebo (P=0.005). The median time to response (platelet count of 50,000/μL or more) in those treated with TAVALISSE®, was two weeks. Rescue medications (including Platelet transfusions and intravenous Immunoglobulin) were required in 26% of responding patients in the TAVALISSE® group and for 45% of patients in the placebo group. Response rates were not influenced by Age, Sex, baseline platelet count, Splenectomy or prior treatment with a TPO-RA. Among patients who had received and later failed to respond to a TPO-RA before enrollment, 17% had a Stable Response to TAVALISSE®. Serious bleeding was not noted in the 29% of patients who achieved a response, whereas it occurred in 5.6% of non-responders and 10.2% of those receiving placebo.
Patients from FIT-1 and FIT-2 trials were also included in an open label expansion cohort (FIT-3). In this study, 23% of those who received placebo in FIT-1 or FIT-2 had a Stable platelet response to TAVALISSE®. The most common toxicities were rash, fatigue, nausea, diarrhea, abdominal pain, hypertension, liver function abnormalities and neutropenia.
It was concluded that TAVALISSE® is the first and only Spleen Tyrosine Kinase (SYK) inhibitor, and by its unique mechanism of action, is an important alternative for patients with difficult to treat chronic ITP. Bussel J, Mayer J, Cervinek L, et al. Treatment of primary adult chronic immune thrombocytopenia (CITP) with fostamatinib, an oral SYK inhibitor: results of two randomized, placebo-controlled phase 3 studies. Abstract #S435. Presented at the 22nd Congress of the European Hematology Association, June 24, 2017; Madrid, Spain.
FDA Approves TAGRISSO® for First-Line Treatment of Metastatic NSCLC
SUMMARY: The FDA on April 19, 2018, approved TAGRISSO® (Osimertinib) for the first-line treatment of patients with metastatic Non Small Cell Lung Cancer (NSCLC), whose tumors have Epidermal Growth Factor Receptor (EGFR) exon 19 deletions or exon 21 L858R mutations, as detected by an FDA-approved test.
Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer. Approximately 10-15% of Caucasian patients and 35-50% of Asian patients with Adenocarcinomas, harbor activating EGFR (Epidermal Growth Factor Receptor) mutations and 90% of these mutations are either exon 19 deletions or L858R point mutations in exon 21.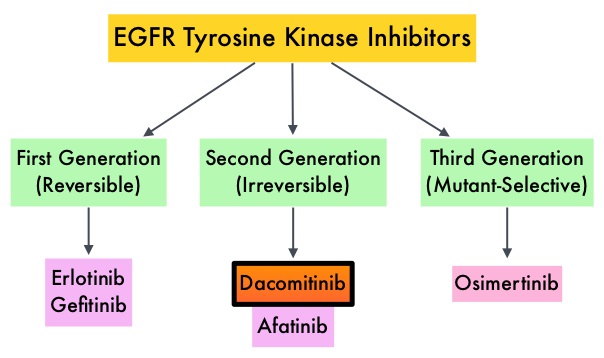 EGFR-Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib), IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), have demonstrated a 60-70% response rate as monotherapy when administered as first line treatment, in patients with metastatic NSCLC, who harbor the sensitizing EGFR mutations. However, majority of these patients experience disease progression within 9-14 months. This resistance to frontline EGFR TKI therapy has been attributed to acquired T790M “gatekeeper” point mutation in EGFR, identified in 50-60% of patients. Put another way, T790M is not relevant in about 40% of patients whose disease progression may be related to other mechanisms.
EGFR-Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib), IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), have demonstrated a 60-70% response rate as monotherapy when administered as first line treatment, in patients with metastatic NSCLC, who harbor the sensitizing EGFR mutations. However, majority of these patients experience disease progression within 9-14 months. This resistance to frontline EGFR TKI therapy has been attributed to acquired T790M “gatekeeper” point mutation in EGFR, identified in 50-60% of patients. Put another way, T790M is not relevant in about 40% of patients whose disease progression may be related to other mechanisms.
TAGRISSO® is a third-generation Epidermal Growth Factor Receptor (EGFR) TKI presently approved by the FDA, for the treatment of patients with metastatic EGFR T790M mutation-positive NSCLC, who had progressed on prior systemic therapy, including an EGFR-TKI. Previously published studies had suggested that TAGRISSO® may also be effective as initial therapy for EGFR mutation-positive advanced NSCLC.
The recent new indication for TAGRISSO® was based on FLAURA, which is a randomized, double blind, phase III clinical trial, conducted to compare the efficacy and safety of first line TAGRISSO® to TARCEVA® or IRESSA® (which are considered standard first line therapies), in NSCLC patients with activating mutations EGFR exon 19 deletions or L858R substitution mutation on exon 21. This study randomized 556 advanced NSCLC treatment naïve patients, with EGFR exon 19 or 21 mutations in a 1:1 ratio, to TAGRISSO® 80 mg orally once daily (N=279) or Standard of Care EGFR-TKI, IRESSA® 250 mg or TARCEVA® 150 mg, orally once daily (N=277). Patients were stratified by mutation status (exon 19 vs 21 mutations) and race (Asian vs non-Asian). Patients with CNS metastases who were neurologically stable, were allowed in this study. The Primary endpoint was Progression Free Survival (PFS).
The median PFS was 18.9 months with TAGRISSO® compared to 10.2 months for the standard therapy (HR=0.46; P<0.001), suggesting a 54% reduction in the risk of disease progression, compared with Standard of Care. TAGRISSO® extended the median Time To Progression by about 9 months. This PFS benefit was consistent across all subgroups of patients, including those with and without CNS metastases at study entry. The Objective Response Rate (ORR) with TAGRISSO® was 80% compared with 76% for TARCEVA® and IRESSA®. The median Duration of Response with TAGRISSO® was 17.2 months versus 8.5 months in the comparator arm. The median Overall Survival was not reached. Grade 3 and 4 toxicities were lower for TAGRISSO® (34%) compared with 45% for TARCEVA® and IRESSA®. Toxicities led to treatment discontinuation for 13% and 18% of patients in the TAGRISSO® and comparator groups, respectively.
It was concluded that TAGRISSO® demonstrated superior efficacy, with a near doubling in median Progression Free Survival, and better tolerability, compared to the Standard of Care, when given as first-line therapy, for patients with advanced EGFR mutation positive NSCLC. Studies are underway, assessing treatments, following resistance to TAGRISSO®.
Osimertinib in Untreated EGFR-Mutated Advanced Non–Small-Cell Lung Cancer. Soria J-C, Ohe Y, Vansteenkiste J, et al. for the FLAURA Investigators. N Engl J Med 2018; 378:113-125
FDA Approves OPDIVO® plus YERVOY® Combination Immunotherapy for intermediate or Poor-risk Advanced Renal Cell Carcinoma
SUMMARY: The FDA on April 16, 2018, granted approvals to OPDIVO® (Nivolumab) and YERVOY® (Ipilimumab) in combination, for the treatment of intermediate or poor-risk, previously untreated advanced Renal Cell Carcinoma (RCC). SUTENT® (Sunitinib) is a MultiKinase Inhibitor (MKI) which simultaneously targets the tumor cell wall, vascular endothelial cell wall as well as the pericyte/fibroblast/vascular/ smooth vessel cell wall and is capable of specifically binding to tyrosine kinases, inhibiting the earlier signaling events and thereby inhibits phosphorylation of VEGF receptor, PDGF receptor, FLT-3 and c-KIT. SUTENT® is the standard first-line intervention for treatment naïve patients with advanced Renal Cell Carcinoma. In a large, multi-center, randomized, phase III study, the median Progression Free Survival (PFS) with SUTENT® was 9.5 months, the Objective Response Rate (ORR) was 25%, and the median Overall Survival was 29.3 months, when compared with Interferon Alfa, in patients with treatment-naïve Renal Cell Carcinoma. This was however associated with a high rate of hematological toxicities.
OPDIVO® is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, whereas YERVOY® is a fully human immunoglobulin G1 monoclonal antibody that blocks Immune checkpoint protein/receptor CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152). Blocking the Immune checkpoint proteins unleashes the T cells, resulting in T cell proliferation, activation and a therapeutic response. OPDIVO® was approved by the FDA in November 2015, for the treatment of advanced RCC in patients who had received prior anti-angiogenic therapy, based on an Overall Survival benefit. YERVOY® is approved for the treatment of metastatic melanoma. Combining OPDIVO® with YERVOY® (Combination immunotherapy) has shown promising efficacy in multiple tumor types, including advanced RCC, with higher Objective Response Rate than either agent alone, and is presently approved for the treatment of advanced malignant melanoma.
This FDA approval was based on CheckMate 214, a randomized open-label phase III trial in which a combination of OPDIVO® plus YERVOY® (N=550) was compared with SUTENT® (N=546), among treatment naïve, clear-cell, advanced Renal Cell Carcinoma (RCC) patients. The authors randomly assigned 1096 patients in a 1:1 ratio to receive OPDIVO® 3 mg/kg IV plus YERVOY® 1 mg/kg IV every 3 weeks for four doses (induction phase) followed by OPDIVO® monotherapy at 3 mg/kg every 2 weeks (maintenance phase) or SUTENT® 50 mg orally once daily for 4 weeks, of each 6-week cycle. Four hundred and twenty five (425) patients in the combination group and 422 patients in the SUTENT® group had intermediate or poor-risk patients. It is estimated that approximately 75% of patients with advanced RCC have intermediate or poor-risk disease and have worse outcomes than those with favorable-risk disease. The coprimary end points were Overall Survival, Objective Response Rate and Progression Free Survival among patients with intermediate or poor prognostic risk disease.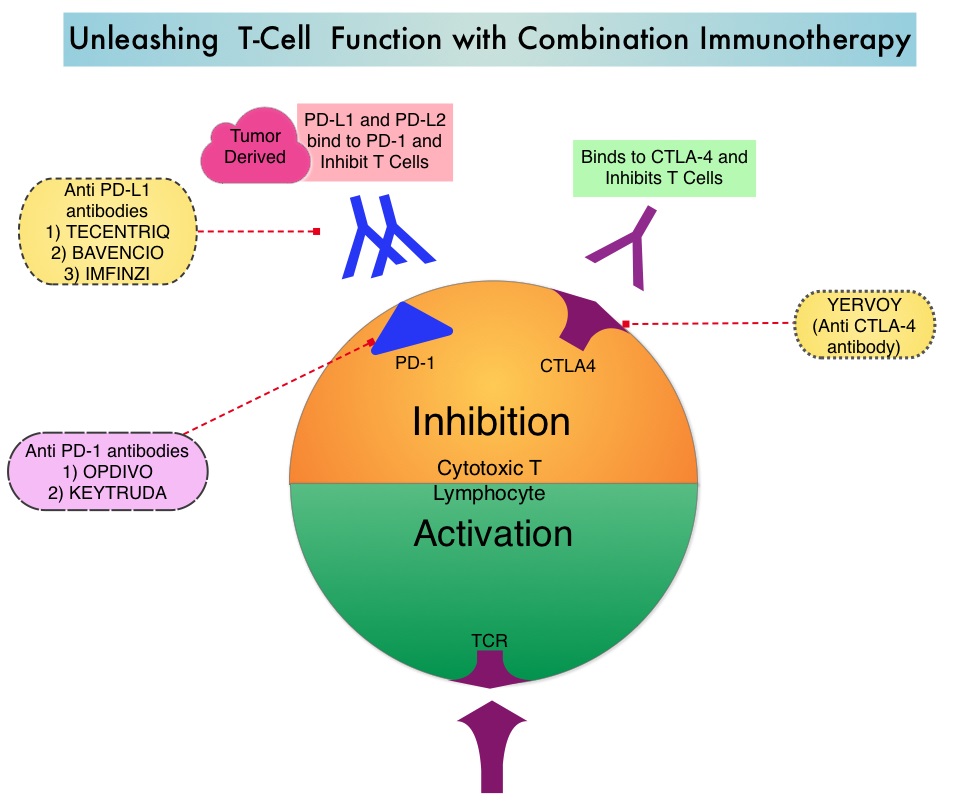
At a median follow-up of 25.2 months, the combination of OPDIVO® and YERVOY® had a significant Overall Survival benefit over SUTENT®. The 18-month Overall Survival rate was 75% with combination immunotherapy and 60% with SUTENT®. The median Overall Survival was not reached with combination immunotherapy versus 26.0 months with SUTENT® (HR=0.63; P<0.001). The Objective Response Rate was 42% with combination immunotherapy versus 27% with SUTENT® (P<0.001), and the Complete Response rate was 9% versus 1% respectively. The median Progression Free Survival was 11.6 months and 8.4 months, respectively but this was not statistically significant per the prespecified threshold. The benefit with combination immunotherapy was not noted in patients with favorable-risk disease. The superior outcomes with combination immunotherapy in patients with intermediate and poor-risk RCC may very well be related to a higher tumor mutational load in this group of patients, compared to those with favorable-risk disease.
In exploratory analyses among 776 intermediate and poor-risk patients, who had quantifiable PD-L1 expression in this study, Overall Survival was longer with Immunotherapy combination compared with SUTENT®, across PD-L1 expression levels. In patients with PD-L1 expression of 1% or greater, the 18-month Overall Survival rate was 81% with combination immunotherapy and 53% with SUTENT®, and the median Overall Survival was not reached versus 19.6 months respectively (HR=0.45). Among patients with PD-L1 expression of 1% or greater, the Objective Response Rate was 58% versus 22% for SUTENT® (P<0.001), the median PFS was 22.8 and 5.9 months, respectively (HR=0.46). A similar trend was noted in patients with PD-L1 expression 5% or greater, as compared with patients with less than 5% PD-L1 expression. Treatment discontinuation, related to adverse events occurred in 22% of the patients in the combination immunotherapy group and 12% in the SUTENT® group.
It was concluded that treatment with a combination of OPDIVO® and YERVOY® resulted in a significantly higher Overall Survival and Objective Response Rates, compared with SUTENT®, among intermediate and poor-risk, previously untreated patients, with advanced Renal Cell Carcinoma. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. Motzer RJ, Tannir NM, McDermott DF, et al. N Engl J Med 2018; 378:1277-1290
Shorter Duration of Adjuvant Chemotherapy for Stage III Colon Cancer
SUMMARY: ColoRectal Cancer (CRC) is the third most common cancer diagnosed in both men and women in the United States. The American Cancer Society estimates that approximately 140,250 new cases of ColoRectal Cancer will be diagnosed in the United States in 2018 and over 50,630 patients are expected to die of the disease. Adjuvant chemotherapy for patients with resected, locally advanced, node-positive (stage III) colon cancer, has been the standard of care since the 1990s. Adjuvant treatment with an ELOXATIN® (Oxaliplatin) based chemotherapy regimen has been considered standard intervention since 2004, for patients with stage III colon cancer, following surgical resection, and has been proven to decrease the chance of recurrent disease. Chemotherapy regimens have included (FOLFOX – Leucovorin, 5-FluoroUracil, ELOXATIN®) or CAPOX/XELOX (XELODA®/Capecitabine and ELOXATIN®), given over a period of 6 months. ELOXATIN® can however be associated with neuropathy which can be long lasting or permanent, depending on the duration of therapy. Additional toxicities with longer duration of chemotherapy include diarrhea, fatigue as well as more office visits.
The IDEA Collaboration is a prospective, pre-planned pooled analysis of 6 concurrently conducted randomized phase III trials, which included 12,834 patients from 12 countries. The goal of this study was to determine if 3 months of adjuvant chemotherapy would be as effective as 6 months of therapy and would be non-inferior. Of the enrolled patients with stage III disease, 13% had T1-2 disease, 66% had T3 tumors and 21% had T4 tumors. Seventy one percent (71%) had N1 disease and 28% of the patients had N2 disease. Approximately 60% had low-risk disease (T1-3, N1) and 40% had high-risk (T4 or N2). Overall, about 40% of patients received CAPOX regimen and 60% received FOLFOX regimen. The primary endpoint was Disease Free Survival (DFS).
At a median follow up of 41.8 months, although non-inferiority of 3 months of therapy as compared with 6 months of therapy could not be confirmed in the overall treatment population, clinically relevant findings according to treatment were noted, in prespecified subgroups of patients. Among those patients who received FOLFOX regimen, 6 months of adjuvant therapy was superior to 3 months (HR=1.16; P=0.001 for superiority of 6-month therapy). However, among those patients who received CAPOX, the Disease Free Survival for 3 months versus 6 months was non-inferior (HR=0.95; P=0.006), and this was highly significant.
In an exploratory analysis, it was noted that among the patient group with low-risk cancers (T1-3, N1 cancers), 3 months of therapy was non-inferior to 6 months of therapy (HR= 1.01) with 3-year disease-free survival of 83.1% and 83.3%, respectively. However, among the patients with high-risk cancers (T4, N2, or both), 6 months of adjuvant therapy was superior to 3 months (HR= 1.12; P=0.01 for superiority).
When subgroup analysis was performed according to treatment and risk group, among the patients with low-risk tumors, 3 months of adjuvant therapy with CAPOX was non-inferior to 6 months of therapy. Outcomes after 3 months of adjuvant FOLFOX therapy were worse than those after 6 months, independent of risk group. For patients with high-risk tumors, 6 months of adjuvant therapy with FOLFOX was superior to 3 months, with a 3-year disease-free survival of 64.7% versus 61.5%. It has been hypothesized that the protracted delivery of a Fluoropyrimidine with CAPOX might have been more effective than the twice-monthly 5-FUinfusions with FOLFOX as an adjuvant therapy. Grade 2 or more neurotoxicity was significantly lower for patients who received 3 months of adjuvant therapy versus 6 months (P <0.0001), regardless of the treatment regimen (17% vs 48% for FOLFOX and 15% vs 45% for CAPOX/XELOX, respectively).
It was concluded by the IDEA collaboration that, a risk-based approach has to be taken when making adjuvant chemotherapy recommendations for patients with stage III colon cancer, taking into consideration choice of treatment regimen and duration of therapy. In patients treated with adjuvant CAPOX/XELOX regimen, 3 months of therapy was as effective as 6 months, particularly in the low risk subgroup. In patients treated with FOLFOX, 6 months of adjuvant therapy compared to 3 months, resulted in a higher rate of Disease Free Survival, particularly in the high-risk subgroup. Duration of Adjuvant Chemotherapy for Stage III Colon Cancer. Grothey A, Sobrero AF, Shields AF, et al. N Engl J Med 2018; 378:1177-1188
AFINITOR® (Everolimus)
The FDA on April 10, 2018 approved AFINITOR® for the adjunctive treatment of adult and pediatric patients aged 2 years and older with Tuberous Sclerosis Complex (TSC)-associated partial-onset seizures. AFINITOR® is also approved for two other manifestations of TSC: TSC-associated SubEpendymal Giant cell Astrocytoma (SEGA) and TSC-associated renal angiomyolipoma. AFINITOR® is a product of Novartis Pharmaceuticals Corp.
RUBRACA® (Rucaparib)
The FDA on April 6, 2018 approved RUBRACA®, a Poly ADP-Ribose Polymerase (PARP) inhibitor, for the maintenance treatment of recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a Complete or Partial Response to platinum-based chemotherapy. RUBRACA® is a product of Clovis Oncology Inc.
BLINCYTO® (Blinatumomab)
The FDA on March 29, 2018 granted accelerated approval to BLINCYTO® for the treatment of adult and pediatric patients with B-cell precursor Acute Lymphoblastic Leukemia (ALL) in first or second complete remission with Minimal Residual Disease (MRD) greater than or equal to 0.1%. BLINCYTO® is a product of Amgen Inc.