SUMMARY: The FDA on May 4, 2018, approved TAFINLAR® (Dabrafenib) and MEKINIST® (Trametinib) in combination, for the treatment of patients with locally advanced or metastatic Anaplastic Thyroid Cancer (ATC) with BRAF V600E mutation, and with no satisfactory locoregional treatment options. ATC is a rare, highly aggressive, undifferentiated malignancy and accounts for 1-2% of all thyroid cancers in the United States and up to 10% of thyroid cancers worldwide. They are all considered Stage IV at diagnosis and are among the most lethal cancers. Despite multimodality therapy with surgery, external beam radiation, and systemic chemotherapy, response rates to standard therapies are less than 15% with a median survival of 5-12 months and a one year Overall Survival of 20-40%.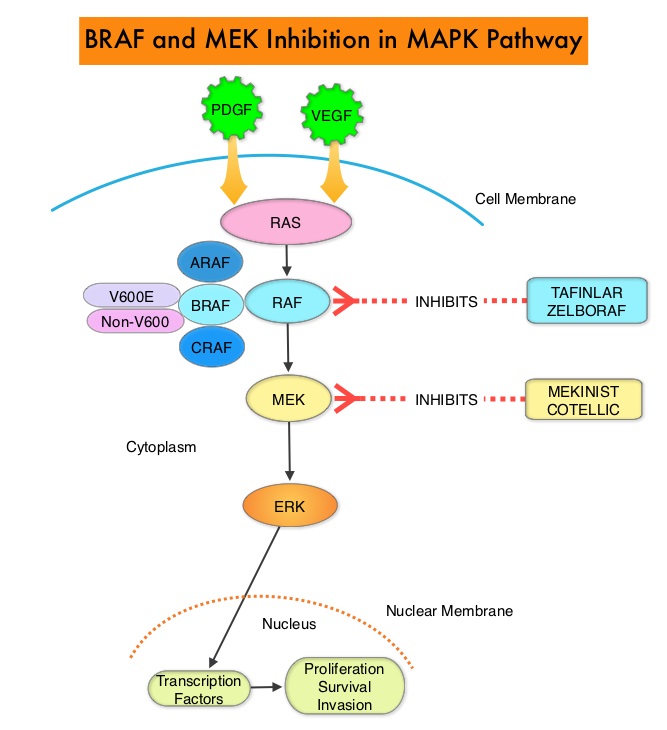
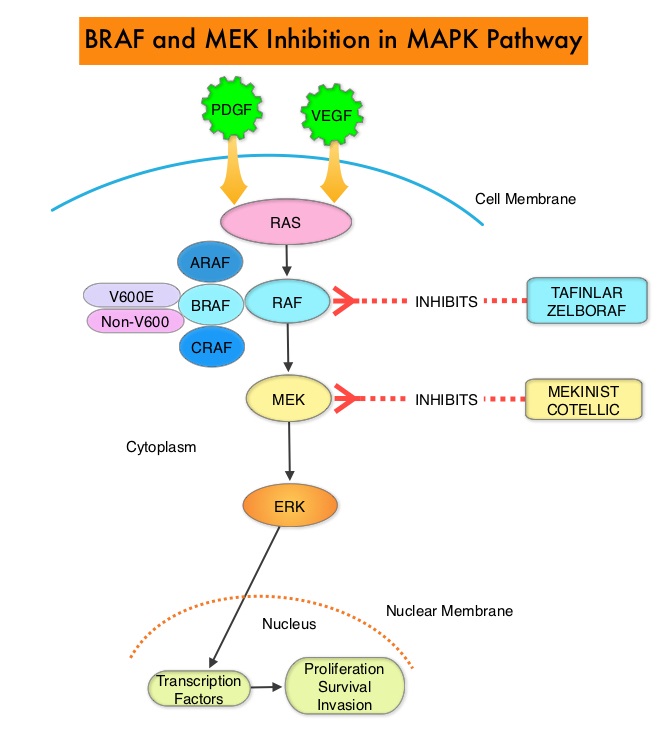
The Mitogen-Activated Protein Kinase pathway (MAPK pathway) is an important signaling pathway which enables the cell to respond to external stimuli. This pathway plays a dual role, regulating cytokine production and participating in cytokine dependent signaling cascade. The MAPK pathway of interest is the RAS-RAF-MEK-ERK pathway. The RAF family of kinases includes ARAF, BRAF and CRAF signaling molecules. BRAF is a very important intermediary of the RAS-RAF-MEK-ERK pathway. BRAF mutations have been demonstrated in 6-8% of all malignancies. Approximately 20-50% of ATCs harbor activating BRAF V600 mutations and in about 50% of ATCs, well-differentiated papillary thyroid cancer precedes or coexists with ATC. It has been postulated that BRAFV600 mutations may be a common and early driver in these well-differentiated thyroid tumors and additional mutations in the course of the disease lead to progressive de-differentiation to ATC. Inhibiting both BRAF and MEK kinases has been shown to enhance antitumor activity and prevent MAPK pathway reactivation, a known resistance mechanism. Combined BRAF plus MEK inhibition has been shown to increase Overall Response Rate (ORR), Duration of Response, Progression Free Survival (PFS), and Overall Survival (OS), compared with BRAF inhibitor monotherapy, in patients with BRAFV600–mutant Melanoma and Non Small Cell Lung Cancer.
This present FDA approval for Anaplastic Thyroid Cancer (ATC) was based on a multicenter, open-label, nonrandomized, phase II trial, which was designed to simultaneously evaluate efficacy and safety of a combination of TAFINLAR® and MEKINIST® in patients with BRAF V600E mutated cancer, in prespecified histologies. A total of 100 patients with BRAF V600E mutated rare cancers in seven prespecified tumor histologies were enrolled. Sixteen (N=16) patients with ATC enrolled in this study were evaluable for response. Patients received TAFINLAR® 150 mg twice daily and MEKINIST® 2 mg once daily, both given orally, until disease progression or unacceptable toxicity. The median patient age was 72 years, 88% had prior surgery, 81% had external beam radiotherapy and 38% had prior chemotherapy. The Primary end point was Overall Response Rate. Secondary end points included Duration of Response, Progression Free Survival, Overall Survival, and safety.
The confirmed Overall Response Rate was 69% and median Duration of Response, Progression Free Survival, and Overall Survival were not reached as a result of ongoing responses at the time of data cut off. Kaplan-Meier estimates at 12 months of Duration of Response, Progression Free Survival, and Overall Survival were 90%, 79%, and 80%, respectively. Responses typically occurred early in the treatment course (within the first 8 weeks of therapy). The most common adverse events were fatigue, pyrexia and nausea and the overall safety profile was similar to previous reports in advanced Metastatic Melanoma and Non Small Cell Lung Cancer.
It was concluded that the combination of TAFINLAR® plus MEKINIST® was well tolerated and is the first regimen to have demonstrated robust clinical activity in BRAF V600E mutated Anaplastic Thyroid Cancer. This is the first FDA approved treatment for patients with this aggressive form of thyroid cancer, and the third malignancy with BRAF V600E gene mutation (others being Melanoma and NSCLC), that this drug combination has been approved to treat. Dabrafenib and Trametinib Treatment in Patients with Locally Advanced or Metastatic BRAF V600–Mutant Anaplastic Thyroid Cancer. Subbiah V, Kreitman RJ, Wainberg AZ, et al. J Clin Oncol. 2018;36:7-13