
Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers and 30% are Squamous Cell Carcinomas (SCC). Non Small Cell Lung Cancer patients with Squamous Cell histology have been a traditionally hard- to-treat, patient group, and less than 15% of patients with advanced Squamous NSCLC survive a year after diagnosis and less than 5% of patients survive for five years or longer. IMpower131 is a multicenter, open-label, phase III study, in which 1021 chemotherapy-naïve patients with stage IV Squamous NSCLC received TECENTRIQ® ((Atezolizumab)) along with Carboplatin, and Paclitaxel, TECENTRIQ® along with Carboplatin, and ABRAXANE® (nab-paclitaxel) or Carboplatin and ABRAXANE® (control group). The addition of TECENTRIQ® to chemotherapy significantly improved median Progression Free Survival across all PD-L1 subgroups. This is the first phase III trial of an immunotherapy-based treatment regimen, to demonstrate a significant improvement in Progression Free Survival, in advanced Squamous NSCLC.
Month: July 2018
Six Months Adjuvant HERCEPTIN® Safer and Efficacious in Early Stage HER2+ Breast Cancer
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 266,120 new cases of invasive breast cancer will be diagnosed in 2018 and about 40,920 women will die of the disease. The HER or erbB family of receptors consist of HER1, HER2, HER3 and HER4. Approximately 15-20% of invasive breast cancers overexpress HER2/neu oncogene, which is a negative predictor of outcomes without systemic therapy. HERCEPTIN® (Trastuzumab) is a humanized monoclonal antibody targeting HER2, and adjuvant chemotherapy given along with HERCEPTIN® reduces the risk of disease recurrence and death, among patients with HER2-positive, early stage breast cancer. The duration of adjuvant HERCEPTIN® therapy has been 12 months and this length of treatment was empirically adopted from the pivotal registration trials.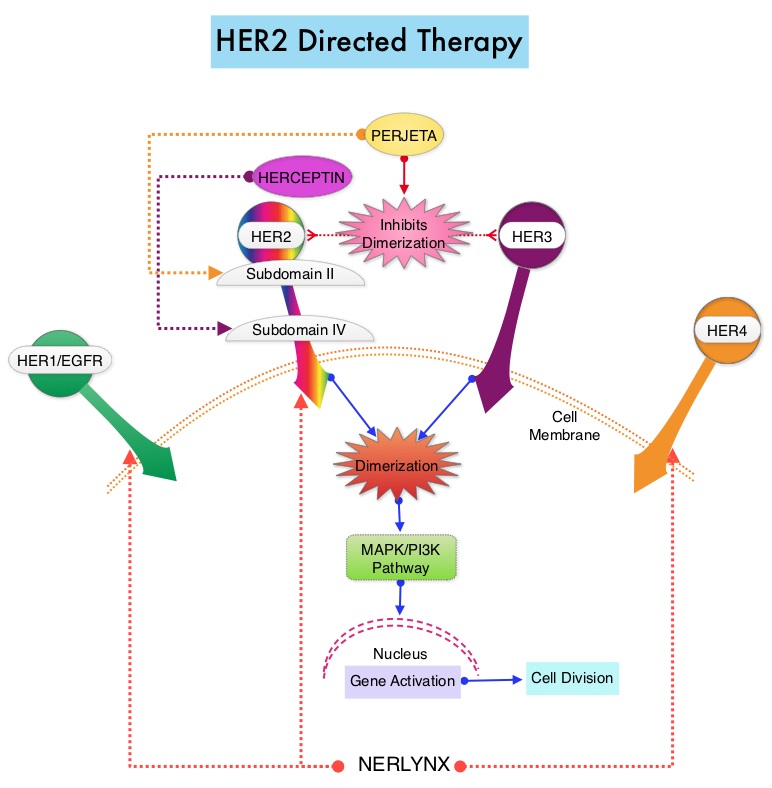
PERSEPHONE is a randomized, phase III, noninferiority trial in which a 6-month course of adjuvant HERCEPTIN® was compared with the standard 12-month course, among patients with HER2-positive early breast cancer. This study was conducted based on the hypothesis that shorter course of treatment with HERCEPTIN® could reduce cardiotoxicities as well as cost without compromising efficacy. This trial randomized 4089 patients across 152 sites in a 1:1 ratio to receive HERCEPTIN® for 6 months (N=2044) or 12 months (N=2045). In this trial, 69% of patients had ER-positive tumors, 41% received Anthracycline-based chemotherapy, 49% received Anthracycline and Taxane-based chemotherapy, 10% received Taxane-based chemotherapy, 85% received adjuvant chemotherapy, and sequential HERCEPTIN® was administered in 54% of patients. This study also included assessment of Left Ventricular Ejection Fraction (LVEF) every 3 months until month 12, as well as continued Quality of Life and health economic assessments at months 18 and 24. The Primary endpoint was Disease Free Survival (DFS) from the time of diagnosis.
At a median follow-up period of 5 years, the 4-year DFS rate was identical in both treatment groups. DFS was 89.8% with 12 months of HERCEPTIN® compared with 89.4% with the 6-month course, which met the criteria for noninferiority (P=0.01). Further, only 4% of the patients enrolled in the 6-month HERCEPTIN® group discontinued HERCEPTIN® treatment due to cardiotoxicities compared with 8% in the 12-month group (P<0.0001), suggesting that the number of patients stopping treatment due to cardiac toxicities was cut in half with the shorter duration of treatment with HERCEPTIN®. Patients receiving shorter course of HERCEPTIN® also had a more rapid recovery of their cardiac LVEF following treatment, compared with the standard of care group (P=0.02).
It was concluded from this largest, reduced duration, noninferiority trial that a shorter 6-month course of adjuvant HERCEPTIN® was noninferior for Disease Free Survival, compared with the standard 12-month schedule, among patients with HER2-positive early breast cancer, with an additional benefit of reduction in cardiac toxicities, as well as cost both to the patients and healthcare systems. Overall Survival data was not available at the time of this analysis. PERSEPHONE: 6 versus 12 months (m) of adjuvant trastuzumab in patients (pts) with HER2 positive (+) early breast cancer (EBC): Randomised phase 3 non-inferiority trial with definitive 4-year (yr) disease-free survival (DFS) results. Earl HM, Hiller L, Vallier A-L, et al. J Clin Oncol 36, 2018 (suppl; abstr 506)
FDA Approves TIBSOVO® for Patients with Relapsed or Refractory Acute Myeloid Leukemia
SUMMARY: The FDA on July 20, 2018, approved TIBSOVO® (Ivosidenib) for adult patients with relapsed or refractory Acute Myeloid Leukemia (AML) with a susceptible Isocitrate DeHydrogenase-1 (IDH1) mutation, as detected by an FDA-approved test. The American Cancer Society estimates that in 2018, 19,520 new cases of Acute Myeloid Leukemia (AML) will be diagnosed in the United States and 10,670 patients will die of the disease. AML can be considered as a group of heterogeneous diseases with different clinical behavior and outcomes. Cytogenetic analysis has been part of routine evaluation when caring for patients with AML. By predicting resistance to therapy, tumor cytogenetics will stratify patients, based on risk and help manage them accordingly. Even though cytotoxic chemotherapy may lead to long term remission and cure in a minority of patients with favorable cytogenetics, patients with high risk features such as unfavorable cytogenetics, molecular abnormalities, prior myelodysplasia and advanced age, have poor outcomes with conventional chemotherapy alone.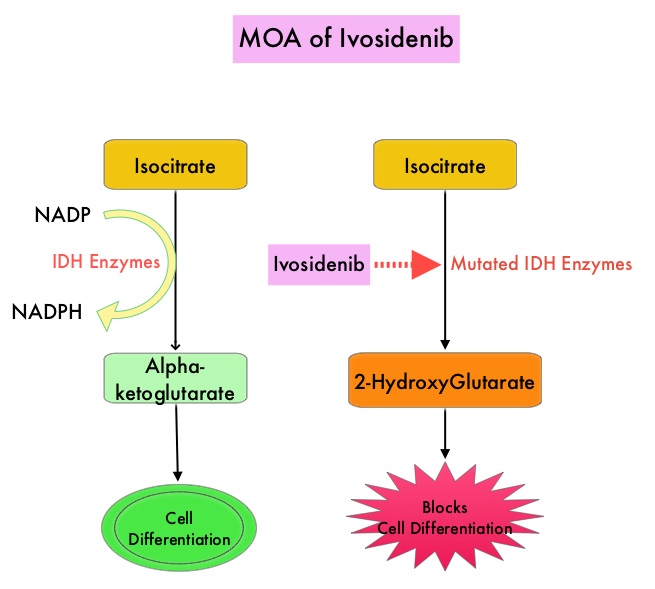
Isocitrate DeHydrogenase (IDH) is a metabolic enzyme that helps generate energy from glucose and other metabolites, by catalyzing the conversion of Isocitrate to Alpha-Ketoglutarate. Alpha-ketoglutarate is required to properly regulate DNA and histone methylation, which in turn is important for gene expression and cellular differentiation. IDH mutations lead to aberrant DNA methylation and altered gene expression thereby preventing cellular differentiation, with resulting immature undifferentiated cells. IDH mutations can thus promote leukemogenesis in Acute Myeloid Leukemia and tumorigenesis in solid tumors and can result in inferior outcomes. There are three isoforms of IDH. IDH1 is mainly found in the cytoplasm, as well as in peroxisomes, whereas IDH2 and IDH3 are found in the mitochondria, and are a part of the Krebs cycle. Approximately 20% of patients with AML, 70% of patients with Low-grade Glioma and secondary Glioblastoma, 50% of patients with Chondrosarcoma, 20% of patients with Intrahepatic cholangiocarcinoma, 30% of patients with Angioimmunoblastic T-cell lymphoma and 8% of patients with Myelodysplastic syndromes/Myeloproliferative neoplasms, are associated with IDH mutations.
TIBSOVO® is an oral, targeted, small-molecule inhibitor of mutant IDH1. IDHIFA® (Enasidenib), an oral, selective, small molecule inhibitor of mutated IDH2 protein was approved in the United States in August 2017 for adult patients with relapsed or refractory AML with an IDH2 mutation. The Complete Remission (CR) rate with the currently available non-targeted therapies for patients with relapsed or refractory AML is 15% or less and the median Overall Survival is less than 4 months, with 30-day mortality of approximately 15% and 60-day mortality of approximately 30%.
The approval of TIBSOVO® was based on an open-label, single arm, multicenter clinical trial that included 174 adult patients with relapsed or refractory AML with an IDH1 mutation. IDH1 mutations were confirmed by the Abbott RealTime® IDH1 assay, the FDA-approved test for selection of patients with AML for treatment with TIBSOVO®. TIBSOVO® was given orally at a starting dose of 500 mg daily until disease progression, unacceptable toxicity, or hematopoietic stem cell transplantation. The median treatment duration was 4.1 months. Twenty one of the 174 patients (12%) received a Stem Cell Transplant following treatment with TIBSOVO®. The median patient age was 67 years, patients had a median number of 2 prior therapies, 87% of the patients had intermediate or poor cytogenetic risk status, 33% had secondary AML and 59% were refractory to their previous therapy. The Primary endpoint was the combined rate of Complete Remission (CR) plus Complete Remission with partial hematologic recovery (CRh) and the rate of conversion from transfusion dependence to independence. CRh was defined as less than 5% of blasts in the bone marrow, no evidence of disease, and partial recovery of peripheral blood counts (platelets more than 50,000/μl and ANC more than 500/μl)
The use of TIBSOVO® was associated with a CR plus CRh rate of 32.8%. The median time to response was 2 months and the median response duration was 8.2 months. The CR and CRh rates were 24.7% and 8.0% respectively. Among the 110 patients who were dependent on Red Blood Cell and/or platelet transfusions at baseline, 37.3% became transfusion independent during any 56-day post-baseline period. Of the 64 patients who were independent of both RBC and platelet transfusions at baseline, 59.4% remained transfusion independent during any 56-day post-baseline period. The most common adverse reactions were fatigue, nausea, diarrhea, rash, pyrexia, arthralgia, leukocytosis and QT prolongation. One effect of the IDH inhibitors are induction of differentiation of the malignant cells, and in 10-20% of patients, a clinical syndrome known as the IDH differentiation syndrome can occur. The IDH differentiation syndrome should be promptly managed by dose interruption and treatment with glucocorticoids, oral hydroxyurea, or both.
It was concluded that in patients with advanced IDH1-mutated relapsed or refractory AML, TIBSOVO® fills an unmet need, with durable molecular remissions and reduction in the need for transfusion support. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. DiNardo CD, Stein EM, de Botton S, et al. N Engl J Med 2018; 378:2386-2398
KISQALI® (Ribociclib)
The FDA on July 18, 2018 expanded the indication for KISQALI® in combination with an Aromatase Inhibitor for pre and perimenopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer, as initial endocrine-based therapy. KISQALI® is a product of Novartis Pharmaceuticals Corporation.
TIBSOVO® (Ivosidenib)
The FDA on July 20, 2018 approved TIBSOVO® for adult patients with relapsed or refractory Acute Myeloid Leukemia (AML) with a susceptible IDH1 mutation, as detected by an FDA-approved test. TIBSOVO® is a product of Agios Pharmaceuticals, Inc.
XTANDI® (Enzalutamide)
The FDA on July 13, 2018 approved XTANDI® for patients with Castration-Resistant Prostate Cancer (CRPC). This approval broadens the indicated patient population to include patients with both non-metastatic CRPC (NM-CRPC) and metastatic CRPC. XTANDI® was previously approved for the treatment of patients with metastatic CRPC. XTANDI® is a product of Astellas Pharma US, Inc.
YERVOY® and OPDIVO®
The FDA on July 10, 2018 granted accelerated approval to YERVOY® (Ipilimumab), for use in combination with OPDIVO® (Nivolumab), for the treatment of patients 12 years of age and older with MicroSatellite Instability-High (MSI-H) or MisMatch Repair deficient (dMMR) metastatic Colorectal Cancer (mCRC), that has progressed following treatment with a Fluoropyrimidine, Oxaliplatin, and Irinotecan. YERVOY® and OPDIVO® are products of Bristol-Myers Squibb Company Inc.
Updated Chronic Lymphocytic Leukemia Guidelines
SUMMARY: The American Cancer Society estimates that for 2018, about 20,940 new cases of Chronic Lymphocytic Leukemia (CLL) will be diagnosed in the US and 4,510 patients will die of the disease. CLL accounts for about 25% of the new cases of leukemia and the average age at the time of diagnosis is around 71 years. B-cell CLL is the most common type of leukemia in adults.
The National Cancer Institute sponsored International Workshop on CLL, issued an update to its consensus guidelines originally published in 2008, prompted by the recent advances in the biology and treatment of patients with CLL. The goal of the updated guidelines is to integrate these new findings into clinical practice and CLL clinical trials.
The following are the major changes or additions reflected in the guidelines
Molecular Genetics
Patients with 17p deletion and TP53 mutations have inferior outcomes and have disease, resistant to standard chemotherapy regimens. These genetic abnormalities can also be acquired over the course of the disease. Patients with CLL should therefore be evaluated for these genetic abnormalities at the time of initial diagnosis and prior to any subsequent line of treatment. These patients have better outcomes when treated with non-chemotherapeutic agents, such as Bruton’s Tyrosine Kinase (BTK) inhibitors, Phosphatidylinositol 3-Kinase (PI3K) inhibitors and BCL2 inhibitors. Even though mutations in NOTCH1 or SF3B1 identified by Next-Generation sequencing have pathogenic as well as prognostic significance, the importance of these mutations has not been validated in prospective trials and their use is therefore not recommended in routine practice.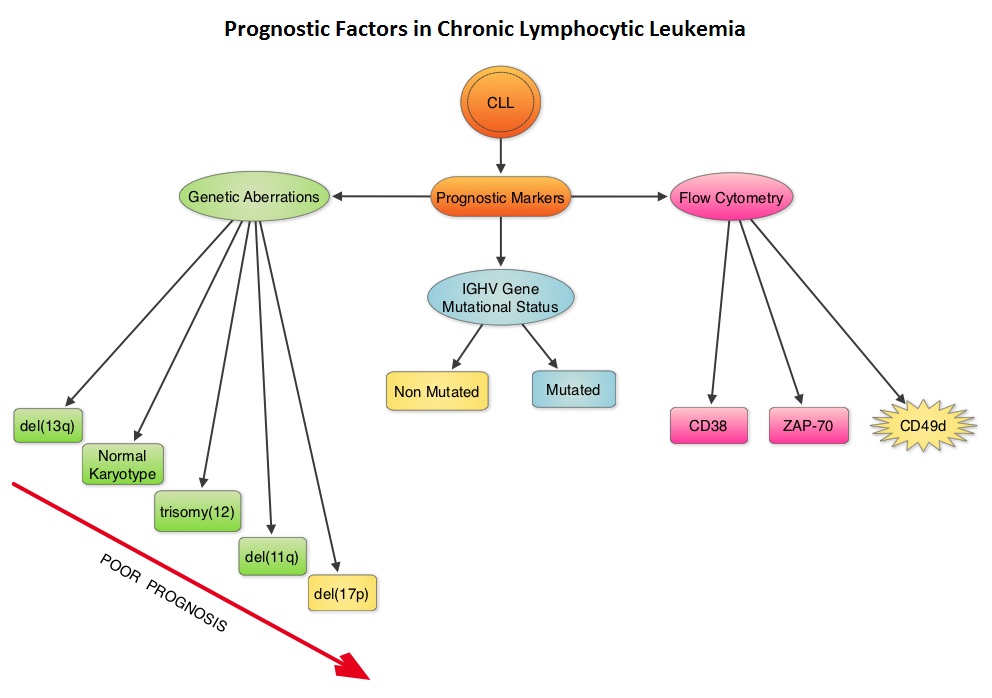
IGHV Mutational status
Retrospective studies have suggested that patients with CLL whose leukemic cells have clonotypically rearranged immunoglobulin genes in germline configuration (Unmutated IGHV gene) demonstrated more aggressive disease and shorter survival time compared to those patients with somatic hypermutations in their IGHV genes (Mutated IGHV gene). The presence of mutated IGHV genes, when combined with additional prognostic factors such as favorable cytogenetics or attainment of a Minimal Residual Disease (MRD) negative state after therapy, characterizes a subgroup of CLL patients with excellent outcome following chemoimmunotherapy with Fludarabine, Cyclophosphamide, and Rituximab. Assessment of IGHV stereotypes however is presently not a part of the routine prognostic work up in CLL.
Serum Biomarkers
Serum markers such as levels of soluble CD23, Thymidine Kinase, and Beta 2-microglobulin are poor prognostic factors and have been shown in several studies to be associated with inferior Overall Survival or Progression Free Survival. Of these, Beta 2-microglobulin has retained independent prognostic value in several multiparameter scores. Assays for these markers should be standardized, and used in prospective clinical trials to validate their relative value in the management of patients with CLL.
Clinical Staging
The two widely accepted staging systems for use in both patient care and clinical trials, the Rai and Binet staging systems rely solely on a physical examination and standard laboratory tests and do not require imaging studies. They are simple and inexpensive and can be readily and consistently applied by physicians worldwide.
Response Definition after Treatment of CLL patients
Assessment of response should include a careful physical examination and evaluation of the blood and bone marrow. For continued therapies or treatment strategies that contain a maintenance phase, the assessment of response should be performed at least 2 months after patients achieve their maximum response or at a time point that is predefined in the protocol and it is not necessary to interrupt therapy for response assessment. The updated guidelines also recommended monitoring for lymphadenopathy, splenomegaly, and hepatomegaly to define relapsed disease and treatment failure, and suggested that the use of imaging in CLL does not typically add much information to the detection of progression or relapse.
MRD Assessment
The desired outcome is complete eradication of the leukemic cells.It was recommended that MRD assessment via multicolor Flow Cytometry, Polymerase Chain Reaction, or Next-Generation sequencing after therapy, be evaluated in the blood and bone marrow, and in clinical trials should be reported with the intent-to-treat population as the denominator and not as a proportion of the responders.
Antiviral Prophylaxis
Patients treated with agents such as Alemtuzumab and Idelalisib (alone or in combination) should be monitored for symptoms or laboratory evidence of opportunistic infections such as Pneumocystis jiroveci or Herpes viridae (Herpes Simplex virus, Varicella-Zoster virus, Cytomegalo virus, Epstein-Barr virus). HBV serological status should be evaluated before treatment with anti-CD20 antibodies as patients may experience reactivation of HBV infections. Appropriate antiviral prophylaxis should be initiated in patients with a history of HBV infection. Infections with JC virus should be ruled out in situations of unclear neurological symptoms, as Progressive Multifocal Leukoencephalopathy has been reported in a few CLL patients treated with anti-CD20 antibodies.
iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Hallek M, Cheson BD, Catovsky D, et al.Blood 2018 131:2745-2760
FDA Approves OPDIVO® and YERVOY® Combination for Metastatic Colorectal Cancer
SUMMARY: The FDA on July 10, 2018, granted accelerated approval to YERVOY® (Ipilimumab) for use in combination with OPDIVO® (Nivolumab), for the treatment of patients 12 years of age and older with MicroSatellite Instability-High (MSI-H) or MisMatch Repair deficient (dMMR) metastatic ColoRectal Cancer, that has progressed following treatment with a Fluoropyrimidine, Oxaliplatin, and Irinotecan. The FDA in July, 2017, granted accelerated approval to single agent OPDIVO® (Nivolumab) for treatment of this same group of patients. ColoRectal Cancer (CRC) is the third most common cancer diagnosed in both men and women in the United States. The American Cancer Society estimates that approximately 140,250 new cases of CRC will be diagnosed in the United States in 2018 and about 50,630 patients are expected to die of the disease. The lifetime risk of developing CRC is about 1 in 21 (4.7%).
The DNA MisMatchRepair (MMR) system is responsible for molecular surveillance and works as an editing tool that identifies errors within the microsatellite regions of DNA and removes them. Defective MMR system leads to MSI (Micro Satellite Instability) and hypermutation, triggering an enhanced antitumor immune response. MSI (Micro Satellite Instability) is therefore a hallmark of defective/deficient DNA MisMatchRepair (dMMR) system and occurs in 15% of all colorectal cancers. Defective MisMatchRepair can be a sporadic or heritable event. Approximately 65% of the MSI tumors are sporadic and when sporadic, the DNA MisMatchRepair gene is MLH1. Defective MisMatchRepair can also manifest as a germline mutation occurring in 1 of the 4 MisMatchRepair genes which include MLH1, MSH2, MSH6, PMS2. This produces Lynch Syndrome (Hereditary Nonpolyposis Colorectal Carcinoma – HNPCC), an Autosomal Dominant disorder and is the most common form of hereditary colon cancer, accounting for 35% of the MSI colorectal cancers. MSI tumors tend to have better outcomes and this has been attributed to the abundance of tumor infiltrating lymphocytes in these tumors from increase immunogenicity. These tumors therefore are susceptible to blockade with immune checkpoint inhibitors. MSI (Micro Satellite Instability) testing is performed using a PCR based assay and MSI-High refers to instability at 2 or more of the 5 mononucleotide repeat markers and MSI-Low refers to instability at 1 of the 5 markers. Patients are considered Micro Satellite Stable (MSS) if no instability occurs. MSI-L and MSS are grouped together because MSI-L tumors are uncommon and behave similar to MSS tumors. Tumors considered MSI-H have deficiency of one or more of the DNA MisMatchRepair genes. MMR gene deficiency can be detected by ImmunoHistoChemistry (IHC). MLH1 gene is often lost in association with PMS2. NCCN Guidelines recommend MMR or MSI testing for all patients with a history of Colon or Rectal cancer.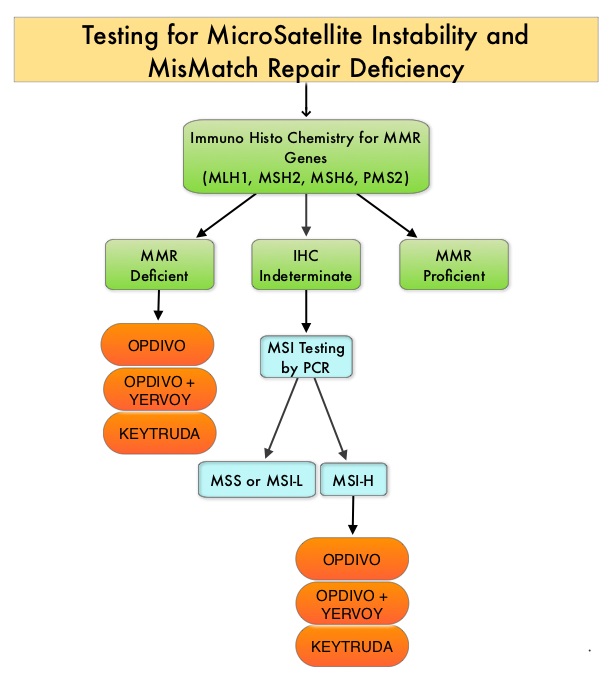
OPDIVO® is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, whereas YERVOY® is a fully human immunoglobulin G1 monoclonal antibody that blocks Immune checkpoint protein/receptor CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152). Blocking the Immune checkpoint proteins unleashes the T cells, resulting in T cell proliferation, activation and a therapeutic response.
This new FDA approval was based on data from the ongoing CheckMate-142, which is a multicenter, open-label, phase II trial, designed to determine within the dMMR/MSI-H population, whether OPDIVO® alone or in combination with another checkpoint inhibitor YERVOY®, could result in meaningful responses in patients with metastatic CRC, following at least one prior line of therapy. This study was not designed to compare the outcomes in cohorts receiving single agent OPDIVO® and combination checkpoint inhibitors. The rationale behind combining OPDIVO® a PD-1 inhibitor and YERVOY®, a CTLA-4 inhibitor, was based on the synergy between these two agents, to promote T-cell antitumor activity, thereby improving upon single-agent activity of OPDIVO®.
This study enrolled 74 patients, who received single agent OPDIVO® 3 mg/kg IV every 2 weeks and 119 patients who received OPDIVO® 3 mg/kg IV plus YERVOY® 1 mg/kg IV, every 3 weeks for four doses, followed by OPDIVO® 3 mg/kg as a single agent every 2 weeks, until unacceptable toxicity or radiographic progression. In this study, from a cohort of 119 patients with MSI-H or dMMR mCRC, 82 patients received prior treatment with a Fluoropyrimidine, Oxaliplatin, and Irinotecan. Among the cohort of 119 patients receiving OPDIVO® plus YERVOY®, the median age was 58 years, 29% had a known history of Lynch syndrome, 24% had BRAF mutations, 37% had KRAS mutations and 22% of patients had high PD-L1 expression (1% or more) on tumor cells at baseline. Primary tumor location was in the right colon in 55% of patients, 25% had left and sigmoid colon disease and 13% had primary tumor in the transverse colon. Seventy six percent (76%) of patients had two or more prior lines of therapy. The Primary end point was investigator-assessed ORR (Overall Response Rate) and Secondary end points included DCR (Disease Control Rate – CR, PR, or stable disease) Safety and tolerability, PFS (Progression Free Survival) and OS (Overall Survival).
At median follow-up of 13.4 months, the ORR in those patients receiving OPDIVO® plus YERVOY®, was 55% and the Disease Control Rate for 12 or more weeks was 80%. Median duration of response was not reached and 94% of the responses were ongoing at data cutoff. PFS and OS rates at 1 year were 71% and 85%, respectively. Further, clinically meaningful and statistically significant improvements were observed in patient-reported outcomes, including functioning, symptoms, and quality of life. Approximately 13% of patients who discontinued treatment because of toxicities still had an ORR of 63%, consistent with that of the overall population. Grade 3 to 4 toxicities occurred in 32% of patients and were manageable. Indirect comparisons in this nonrandomized CheckMate-142 trial suggested that OPDIVO® plus YERVOY® provided numerically higher response rates and improved long term clinical benefit relative to OPDIVO® monotherapy.
It was concluded that OPDIVO® plus YERVOY® results in a high response rates, encouraging PFS and OS at 12 months, with manageable toxicities. These data from the CheckMate-142 study support the use of OPDIVO® as a single agent or in combination with YERVOY®, for the treatment of patients with previously treated dMMR/MSI-H metastatic CRC. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair–Deficient/Microsatellite Instability–High Metastatic Colorectal Cancer. Overman MJ, Lonardi S, Wong KYM, et al. J Clin Oncol. 2018;36:773-779
Late Breaking Abstract – ASCO 2018 Blood Test Demonstrates High Specificity for Detection of Early Stage Lung Cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Survival rates however are significantly higher when lung cancer is diagnosed early. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas, and 10% are Large cell carcinomas.
Although the U.S. Preventive Services Task Force (USPSTF) has recommended annual screening for lung cancer with Low-Dose Computed Tomography (LDCT) for individuals with significant smoking history, screening is vastly underutilized, with a screening rate of less than 2% among smokers eligible for screening. Screening for lung cancer using a peripheral blood sample may improve lung cancer screening rates. Analysis of cell-free DNA (cfDNA) from peripheral blood (Liquid Biopsy), is presently approved to select EGFR targeted therapies (cobas EGFR mutation test), in patients with advanced Non Small Cell Lung Cancer. However, the role of cell-free DNA analysis for early detection of lung cancer is not well established.
The Circulating Cell-Free Genome Atlas (CCGA) is a prospective, multi-center, observational study and is the largest study ever initiated, to develop a noninvasive, liquid biopsy assay for early cancer detection, based on cell-free DNA (cfDNA). This study has currently enrolled 10,012 of a planned 15,000 participants, including people with a recent cancer diagnosis and also a control group of individuals with no known malignancy (70% with cancer, 30% without cancer), across 141 sites in the United States and Canada. This report is one of the first pre-planned sub-studies from the CCGA, involving investigation of blood samples from 1,627 participants (878 patients with newly diagnosed untreated cancer including 127 patients with lung cancer and 749 controls – 580 controls and 169 technical assay controls ), across 20 tumor types and all clinical stages.
The cell-free DNA was isolated from the peripheral blood and analyzed using the following three sequencing methods that were designed to detect cancer-defining signals (mutations and other genomic changes), that could be utilized for early cancer detection.
Targeted sequencing to detect somatic (non-inherited) mutations, such as Single Nucleotide Variants and small insertions and/or deletions, in specific sections of the genome.
Whole-Genome Sequencing (WGS) to detect somatic gene copy number changes across the genome.
Whole-Genome Bisulfite Sequencing (WGBS) of cfDNA to detect abnormal patterns of cfDNA methylation (epigenetic changes)
In this initial sub-study, the authors explored the ability of the above three different assays to detect cancer in 127 people with stage I-IV lung cancer. It was noted that biologic signals suggesting lung cancer were detected and comparable across all assays, and the signal increased with cancer stage. At 98% specificity, the Targeted sequencing detected 51% of early-stage (stage I-IIIA) lung cancers and 89% of late-stage (stage IIIB-IV) lung cancers. Whole-Genome Sequencing detected 38% of early-stage cancers and 87% of late-stage cancers. Whole-Genome Bisulfite Sequencing had similar efficacy, detecting 41% of early stage lung cancers and 89% of late-stage cancers. Similar sensitivities were noted across all assays for adenocarcinoma, squamous cell and small cell lung cancer. False positive rates were low. Of the 580 control participants without cancer at study enrollment, less than 1% (five participants) had cancer-like signal across all three assays, of whom two were subsequently diagnosed with cancer. This highlights the potential for these assays to detect early stage cancers. The authors caution that a large proportion of cell-free DNA is derived from White Blood Cells (WBCs) and DNA mutations in the WBC population can also be generated by processes other than cancer such as clonal hematopoiesis during human aging. In this study, signal generated from the WBCs was subtracted resulting in a cleaner signal, only from tumor related variants.
It was concluded that based on the initial results from the CCGA study, it is possible to detect early-stage lung cancer, with a high degree of specificity, from a simple blood test, using genome sequencing. The authors plan to further optimize the assays and validate results in a larger group of people. Genome-wide sequencing for early stage lung cancer detection from plasma cell-free DNA (cfDNA): The Circulating Cancer Genome Atlas (CCGA) study. Oxnard GR, Maddala T, Hubbell E, et al. J Clin Oncol. 2018;36(suppl; abstr LBA8501)