SUMMARY: The FDA on May 24, 2019 approved JAKAFI® (Ruxolitinib) for steroid-refractory acute Graft-Versus-Host Disease (GVHD) in adult and pediatric patients 12 years and older. Acute GVHD is a frequent and severe inflammatory complication of allogeneic Hematopoietic Cell Transplantation (HCT), and is a reaction of donor immune cells against host tissues. It is estimated that in the US over 8000 patients undergo allogeneic HCT each year and about 35-50% of recipients will develop acute GVHD, which remains a significant cause of morbidity and mortality in allogeneic HCT recipients. Following the preparative regimen, a series of inflammatory reactions lead to damage to the host epithelial cells by activated donor T cells. GVHD can be acute or chronic, with acute GVHD typically occurring within the first 100 days following an allogeneic transplant. Approximately 40% of patients with acute GVHD have severe disease, with a one year survival of 50% or less. Acute GVHD typically involves the skin, often starting in the palms and soles (rash/dermatitis), liver (hepatitis/jaundice), and gastrointestinal tract (abdominal pain/diarrhea). Acute GVHD is a clinical diagnosis, although histologic confirmation may be extremely helpful, if the symptoms and presentation are atypical.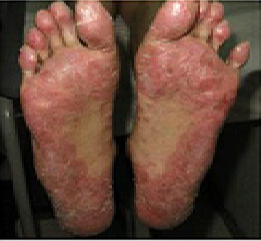 Risk factors for the development of acute GVHD include degree of HLA disparity, gender disparity, increased age of both the recipient and the donor, multiparous female donors, ineffective GVHD prophylaxis, intensity of the transplant conditioning regimen and the source of graft (peripheral blood or bone marrow greater than umbilical cord blood).
Risk factors for the development of acute GVHD include degree of HLA disparity, gender disparity, increased age of both the recipient and the donor, multiparous female donors, ineffective GVHD prophylaxis, intensity of the transplant conditioning regimen and the source of graft (peripheral blood or bone marrow greater than umbilical cord blood).
Patients with acute GVHD are often treated by optimizing their immunosuppression and adding methylprednisolone, with approximately 50% of patients responding to this intervention. If symptoms do not improve after a week or if progression is noted after 3 days of treatment, patients receive salvage immunosuppressive intervention, since no standard treatment with meaningful benefit has been identified.
JAKAFI® (Ruxolitinib) is a potent JAK1 and JAK2 inhibitor and exerts its mechanism of action by targeting and inhibiting the dysregulated JAK2-STAT signaling pathway. JAKAFI® in animal models was shown to reduce IL-1β, IL-6, or IFN-γ and TNF and other cytokines implicated in lymphocyte activation characteristic of GVHD.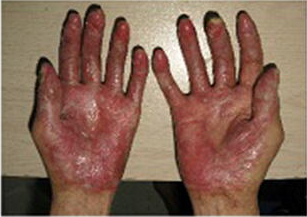 In previously published studies, JAKAFI® when used in patients with refractory GVHD demonstrated an Overall Response Rate of 85% in acute or chronic GVHD, with a 25% Complete Remission rate.
In previously published studies, JAKAFI® when used in patients with refractory GVHD demonstrated an Overall Response Rate of 85% in acute or chronic GVHD, with a 25% Complete Remission rate.
The present FDA approval was based on data from REACH1 study, which is an open-label, single-arm, multicenter, phase II trial of JAKAFI® in combination with corticosteroids, in patients with steroid-refractory grade II-IV acute GVHD. Of the 71 patients enrolled in this study, 49 patients were refractory to steroids alone, 12 patients had received two or more prior therapies for GVHD and 10 patients did not otherwise meet the FDA definition of steroid-refractory state. JAKAFI® was administered at 5 mg orally twice daily, and the dose could be increased to 10 mg twice daily after three days, in the absence of toxicity.
The Primary endpoint of this trial was the Day 28 Overall Response Rate (ORR), defined as a Complete Response (CR), Very Good Partial Response (VGPR) or Partial Response (PR), based on the Center for International Blood and Marrow Transplant Research (CIBMTR) criteria. The Day 28 ORR in the 49 patient’s refractory to steroids alone was 57% with a CR rate of 31%. The most frequently reported adverse reactions were infections (55%) and edema (51%), and the most common laboratory abnormalities were anemia, thrombocytopenia and neutropenia.
It was concluded that for patients with acute GVHD who do not adequately respond to steroids, therapies are limited and JAKAFI® is a new treatment option that fulfills this unmet need. Results from REACH1, a Single-Arm Phase 2 Study of Ruxolitinib in Combination with Corticosteroids for the Treatment of Steroid-Refractory Acute Graft-Vs-Host Disease. Jagasia M, Perales M-A, Schroeder MA, et al. Blood 2018 132:601; doi: https://doi.org/10.1182/blood-2018-99-116342