
The FDA on May 10, 2019 approved CYRAMZA® as a single agent for HepatoCellular Carcinoma (HCC) in patients who have an Alpha FetoProtein (AFP) of 400 ng/mL or more, and have been previously treated with NEXAVAR® (Sorafenib). CYRAMZA® is a product of Eli Lilly and Company.
Month: May 2019
FDA Approves KADCYLA® for Early Breast Cancer
SUMMARY: The FDA on May 3, 2019, approved KADCYLA® (Ado-Trastuzumab Emtansine) for the adjuvant treatment of patients with HER2-positive early breast cancer, who have residual invasive disease after neoadjuvant Taxane and HERCEPTIN® (Trastuzumab)-based treatment. Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 268,600 new cases of invasive breast cancer will be diagnosed in 2019 and about 41,760 women will die of the disease. The HER or erbB family of receptors consist of HER1, HER2, HER3 and HER4. Approximately 15-20% of invasive breast cancers overexpress HER2/neu oncogene, which is a negative predictor of outcomes without systemic therapy. HERCEPTIN® (Trastuzumab) is a humanized monoclonal antibody targeting HER2, and adjuvant and neoadjuvant chemotherapy given along with HERCEPTIN® reduces the risk of disease recurrence and death, among patients with HER2-positive, early stage as well as advanced metastatic breast cancer. Since the approval of HERCEPTIN®, several other HER2-targeted therapies have become available. The duration of adjuvant HERCEPTIN® therapy has been 12 months and this length of treatment was empirically adopted from the pivotal registration trials. 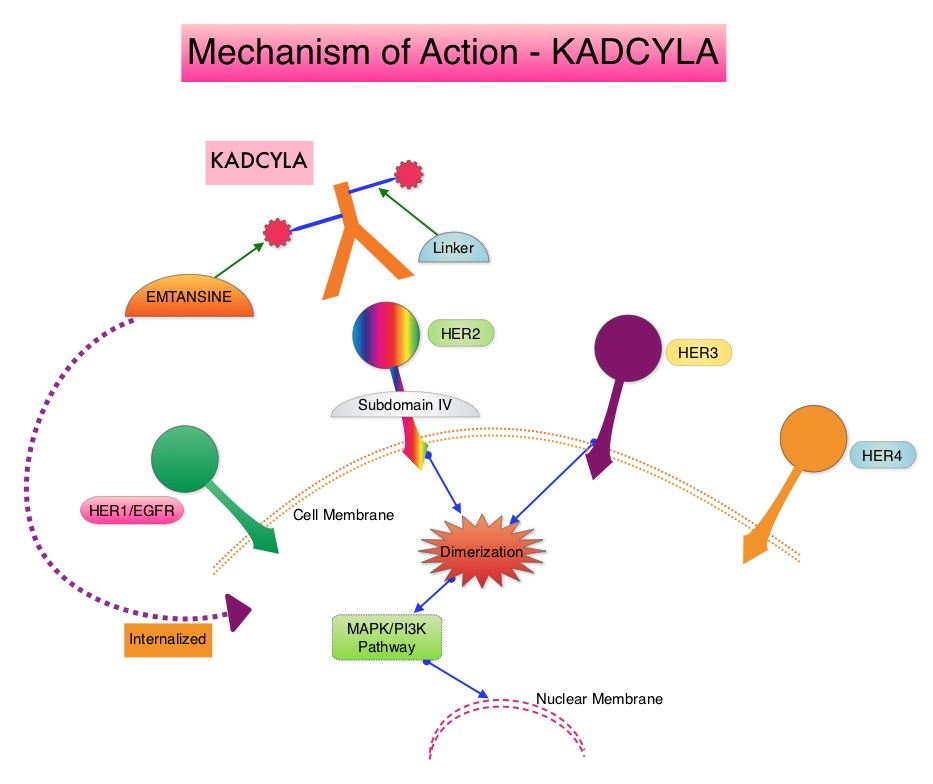
KADCYLA® is an Antibody-Drug Conjugate (ADC) comprised of the antibody HERCEPTIN® and the chemotherapy agent Emtansine, linked together. Upon binding to the HER2 receptor, it not only inhibits the HER2 signaling pathways but also delivers a chemotherapy agent Emtansine, a microtubule inhibitor, directly inside the tumor cells. This agent is internalized by lysosomes and destroys the HER2-positive tumor cells upon intracellular release. In the EMILIA trial, KADCYLA® was associated with significant increase in Overall Survival when compared with TYKERB® (Lapatinib) plus XELODA® (Capecitabine), in HER2-positive metastatic breast cancer patients, who had previously received HERCEPTIN® and a Taxane.
It is well established that patients with HER2-positive early breast cancer following HERCEPTIN® based neoadjuvant therapies have a pathological Complete Response rate of 40-60%. Those without a pathological Complete Response tend to have significantly less favorable outcomes. These patients irrespective of pathological response status complete their standard adjuvant therapy which includes 12 months of HER2-targeted therapy. KATHERINE trial was conducted to evaluate the benefit of switching from standard HER2-directed therapy to single-agent KADCYLA®, after neoadjuvant chemotherapy along with either single or dual HER2 targeted therapy, in patients with residual invasive cancer at surgery. This study was conducted to address the unmet need of patients who have residual invasive breast cancer after receiving neoadjuvant chemotherapy plus HER2-targeted therapy.
The KATHERINE trial is an open-label, phase III global study, which compared KADCYLA® with HERCEPTIN®, as an adjuvant treatment for patients with HER2-positive early breast cancer, who had residual invasive disease following neoadjuvant chemotherapy and HERCEPTIN®. This study included 1,486 patients with HER2-positive early stage breast cancer, who were found to have residual invasive disease in the breast or axillary lymph nodes at surgery, following at least six cycles (16 weeks) of neoadjuvant chemotherapy with a Taxane (with or without Anthracycline) and HERCEPTIN®. Within 12 weeks of surgery, patients (N=1486) were randomly assigned in a 1:1 ratio to KADCYLA® 3.6 mg/kg IV every 3 weeks or HERCEPTIN® 6 mg/kg IV every 3 weeks, for 14 cycles (743 patients in each group). Both treatment groups were well balanced and hormone receptor positive disease was present in 72% of the patients. The majority of the patients (77%) had received an Anthracycline-containing neoadjuvant chemotherapy regimen, and in 19% of the patients, another HER2-targeted agent in addition to HERCEPTIN® (dual HER2 blockade) had been administered as a component of neoadjuvant therapy. The Primary end point was invasive Disease Free Survival (defined as freedom from ipsilateral invasive breast tumor recurrence, ipsilateral locoregional invasive breast cancer recurrence, contralateral invasive breast cancer, distant recurrence, or death from any cause). The median duration of follow up was 41.4 months in the KADCYLA® group and 40.9 months in the HERCEPTIN® group.
At the prespecified interim analysis, invasive disease occurred in 12.2% of patients who received KADCYLA® and 22.2% of patients who received HERCEPTIN®. The estimated percentage of patients who were free of invasive disease at 3 years was 88.3% in the KADCYLA® group and 77.0% in the HERCEPTIN® group. Invasive Disease Free Survival, which was the Primary end point of the study, was significantly higher in the KADCYLA® group than in the HERCEPTIN® group (HR=0.50; P<0.001).This suggested that KADCYLA® reduced the risk of developing an invasive breast cancer recurrence or death by 50%. Distant recurrence as the first invasive disease event occurred in 10.5% of patients in the KADCYLA® group and in 15.9% of the HERCEPTIN® group. A consistent benefit was seen across all prespecified subgroups. Adverse events were consistent with the known safety profile of KADCYLA®, with more toxicities associated with KADCYLA® than with HERCEPTIN®. Additional follow-up will be necessary to determine the Overall Survival benefit with adjuvant KADCYLA®.
It was concluded that among patients with HER2-positive early breast cancer who had residual invasive disease after completion of neoadjuvant therapy, substituting KADCYLA® for adjuvant HERCEPTIN® reduced the risk of recurrence of invasive breast cancer or death by 50%, with the benefit seen across all patient subgroups. The authors added that even though KATHERINE trial focused on higher-risk patients with residual invasive breast cancer after completion of neoadjuvant chemotherapy, CNS recurrence remains a persistent problem. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. von Minckwitz G, Huang C-S, Mano MS, et al. for the KATHERINE Investigators. N Engl J Med 2019;380:617-628
Concomitant Use of Direct Oral Anticoagulants with Antiplatelet Agents Associated with Lower Risk of Major Bleeding Compared with Concomitant Warfarin and Antiplatelet Agents
SUMMARY: It is estimated that up to 30% of patients with nonvalvular atrial fibrillation may receive antiplatelet agents along with oral anticoagulants, due to comorbid cardiovascular conditions. The concomitant use of Vitamin K Antagonist (VKA) such as Warfarin along with antiplatelet agents, has in previously published studies, shown to increase the risk of bleeding, compared with VKAs alone.
Direct Oral AntiCoagulants (DOACs) are often prescribed for thromboembolic events. This class of anticoagulants, have a rapid onset and offset of action, short half-life, predictable anticoagulant effects, no laboratory monitoring and fixed dosing schedule. The half-life of these agents can however be prolonged in those with renal insufficiency and may be unsafe and DOACs are ineffective in patients with mechanical heart valves. Direct Oral AntiCoagulants have a favorable efficacy and safety profile, compared with Vitamin K Antagonists (VKAs) and are increasingly being used for ischemic stroke prevention among patients with nonvalvular atrial fibrillation. In several clinical studies, DOACs have been shown to reduce the rate of major bleeding by 28% and the rates of intracranial and fatal hemorrhage by 50%, when compared to Vitamin K Antagonist (VKA) such as Warfarin. Meta-analysis of randomized controlled trials (RCTs) assessing the efficacy of DOACs along with AcetylSalicylic Acid (ASA), in nonvalvular atrial fibrillation has shown similar risk of major bleeding but a decreased risk of intracranial hemorrhage, when compared with VKAs plus ASA. Some of the studies included in this meta-analysis however had methodological limitations.
In order to address this clinically important safety issue, the authors conducted this population-based study to compare the incidence of intracranial hemorrhage, gastrointestinal bleeding, and other major bleeding between concomitant DOAC/antiplatelet use and concomitant VKA/antiplatelet use, in patients with nonvalvular atrial fibrillation. This study was conducted among a cohort of patients with newly diagnosed nonvalvular atrial fibrillation, between January 2011 and March 2014, using computerized health care databases from Quebec. Of the 14, 407 patients included in this study, 5301 patients initiated concomitant DOAC/antiplatelet use, while 9106 patients initiated concomitant VKA/antiplatelet use. DOACs included PRADAXA® (Dabigatran), XARELTO® (Rivaroxaban), or ELIQUIS® (Apixaban) and antiplatelet agents included ASA (Aspirin), Dipyridamole, PLAVIX® (Clopidogrel), EFFIENT® (Prasugrel), or BRILINTA® (Ticagrelor). Three separate analyses were conducted for intracranial hemorrhage, gastrointestinal bleeding, and other major bleeding. The median follow up was 1.6 months which was primarily driven by discontinuation of antiplatelet therapy.
It was noted that concomitant DOAC/antiplatelet therapy was associated with a similar risk of gastrointestinal bleeding (HR 1.08) but with a decreased risk of intracranial hemorrhage (HR 0.46) and other major bleeding (HR 0.68), compared with concomitant VKA/antiplatelet therapy.
The authors concluded that based on the results of this population-based study, compared with concomitant Vitamin K Antagonist /antiplatelet use, concomitant Direct Oral AntiCoagulants/antiplatelet use was associated with a similar risk of gastrointestinal bleeding, but a lower risk of intracranial hemorrhage and other major bleeding. These findings could provide guidance to physicians and help in decision making, for patients requiring concomitant treatment with oral anticoagulants and antiplatelets. Concomitant Use of Direct Oral Anticoagulants with Antiplatelet Agents and the Risk of Major Bleeding in Patients with Nonvalvular Atrial Fibrillation. Douros A, Renoux C, Yin H, et al. The American Journal of Medicine 2019; 132:191-199
KADCYLA® (Ado-trastuzumab Emtansine)
The FDA on May 3, 2019, approved KADCYLA® for the adjuvant treatment of patients with HER2-positive early breast cancer (EBC), who have residual invasive disease after neoadjuvant Taxane and HERCEPTIN® (trastuzumab)-based treatment.KADCYLA® is a product of Genentech, Inc.
Patients should be selected based on an FDA-approved companion diagnostic for KADCYLA®. FDA also approved both the Ventana Medical Systems, Inc. PATHWAY anti-HER-2/neu (4B5) Rabbit Monoclonal Primary Antibody assay and the INFORM HER2 Dual ISH DNA Probe Cocktail assay as companion diagnostic devices for selecting patients.
Six Prognostic Factors That Predict Invasive Breast Cancer Recurrence after DCIS
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 268,600 new cases of female breast cancer will be diagnosed in 2019 and about 41,760 women will die of the disease. Carcinoma in situ of the breast also known as Ductal Carcinoma In Situ (DCIS) is defined as a malignant proliferation of ductal epithelial cells that are confined to the milk ducts without invasion of the basement membrane, and is a precursor lesion to invasive carcinoma. DCIS accounts for approximately 25% of all newly diagnosed breast cancers. Patients with small, screening-detected lesions, are often treated with breast-conserving surgery (to prevent the development of invasive breast cancer), followed by adjuvant radiation and hormonal therapy, although neither of the latter two interventions have been shown to improve survival outcomes. As such, a significant number of patients are overtreated. There remains a large unmet need, to distinguish relatively benign DCIS from DCIS that will develop into invasive breast cancer.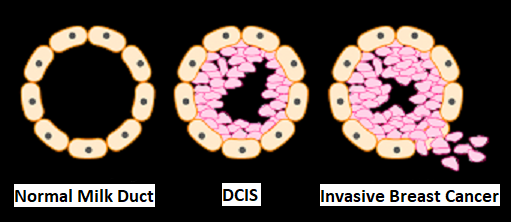
The authors in this study performed a systematic review from 1970 to 2018, with meta-analyses of 1,781 articles from the PubMed database, to summarize current knowledge on prognostic factors for invasive disease, after a diagnosis of DCIS. The number of patients in the included studies ranged from 52 to 37,692. Of all the articles reviewed, 40 articles met the inclusion criteria. Eligible studies assessed risk of invasive recurrence in women primarily diagnosed and treated for DCIS, and included at least 10 ipsilateral-invasive breast cancer events and 1 year of follow up. The mean follow up time ranged from 3.2 to 15.8 years. Quality in Prognosis Studies (QUIPS) tool was used for risk-of-bias assessment (A working group comprising epidemiologists, statisticians, and clinicians developed this tool based on previous research and this tool can inform judgements of risk-of-bias in prognostic research). Meta-analyses were performed to estimate the average effect size of the prognostic factors.
The researchers identified six prognostic factors in the meta-analyses that were statistically significant and were associated with a 36% to 84% increase in the relative risk of recurrence of invasive disease after a DCIS diagnosis. These six factors included-
1) African American race (43% higher risk)
2) Premenopausal status (59% higher risk)
3) Detection by palpation (84% higher risk)
4) Positive margins (63% higher risk)
5) High histologic grade (36% higher risk)
6) High p16 expression (51% higher risk).
Further, the authors identified frequently occurring biases in studies on invasive recurrence after DCIS and the highest risk of bias was attributable to insufficient handling of confounders and poorly described study groups. They added that avoiding these common methodological pitfalls can improve future study designs.
It was concluded that this study results may help clinicians distinguish indolent from potentially aggressive DCIS and prevent overtreatment.
Predictors of an Invasive Breast Cancer Recurrence after DCIS: A Systematic Review and Meta-analyses. Visser LL, Groen EJ, van Leeuwen FE, et al. Cancer Epidemiol Biomarkers Prev. 2019;28:835-845
FDA Approves TIBSOVO® for Newly Diagnosed AML Patients Ineligible for Intensive Chemotherapy
SUMMARY: The FDA on May 2, 2019, approved TIBSOVO® (Ivosidenib) for newly-diagnosed acute myeloid leukemia (AML) with a susceptible IDH1 (Isocitrate DeHydrogenase-1) mutation, as detected by an FDA-approved test, in patients who are at least 75 years old or who have comorbidities that preclude the use of intensive induction chemotherapy. The American Cancer Society estimates that for 2019, 21,450 new cases of Acute Myeloid Leukemia (AML) will be diagnosed in the United States and 10,920 patients will die of the disease. AML can be considered as a group of heterogeneous diseases with different clinical behavior and outcomes. Cytogenetic analysis has been part of routine evaluation when caring for patients with AML. By predicting resistance to therapy, tumor cytogenetics will stratify patients, based on risk and help manage them accordingly. Even though cytotoxic chemotherapy may lead to long term remission and cure in a minority of patients with favorable cytogenetics, patients with high risk features such as unfavorable cytogenetics, molecular abnormalities, prior Myelodysplasia and advanced age, have poor outcomes with conventional chemotherapy alone.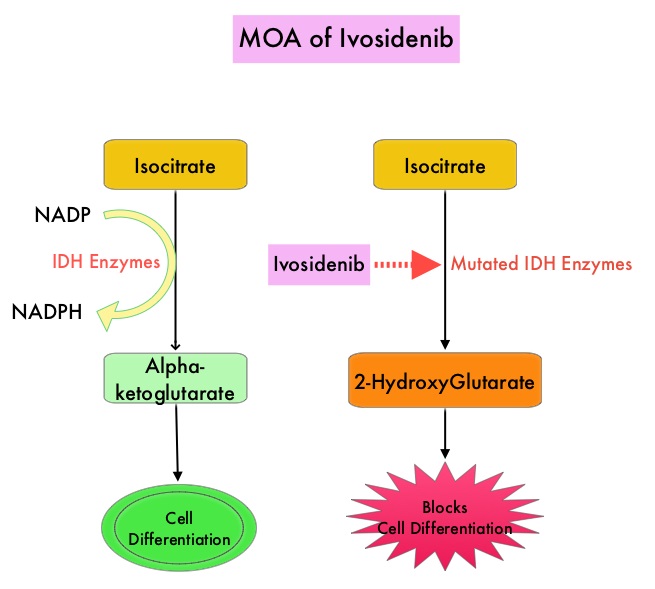
Isocitrate DeHydrogenase (IDH) is a metabolic enzyme that helps generate energy from glucose and other metabolites, by catalyzing the conversion of Isocitrate to Alpha-Ketoglutarate. Alpha-ketoglutarate is required to properly regulate DNA and histone methylation, which in turn is important for gene expression and cellular differentiation. IDH mutations lead to aberrant DNA methylation and altered gene expression thereby preventing cellular differentiation, with resulting immature undifferentiated cells. IDH mutations can thus promote leukemogenesis in Acute Myeloid Leukemia and tumorigenesis in solid tumors and can result in inferior outcomes. There are three isoforms of IDH. IDH1 is mainly found in the cytoplasm, as well as in peroxisomes, whereas IDH2 and IDH3 are found in the mitochondria, and are a part of the Krebs cycle. Approximately 20% of patients with AML, 70% of patients with Low-grade Glioma and secondary Glioblastoma, 50% of patients with Chondrosarcoma, 20% of patients with Intrahepatic cholangiocarcinoma, 30% of patients with Angioimmunoblastic T-cell lymphoma and 8% of patients with Myelodysplastic syndromes/Myeloproliferative neoplasms, are associated with IDH mutations. TIBSOVO® is an oral, targeted, small-molecule inhibitor of mutant IDH1. The FDA in July, 2018, approved TIBSOVO® for adult patients with relapsed or refractory AML with a susceptible IDH1 mutation.
The present first line approval by the FDA was based on an open-label, single-arm, multicenter clinical trial (Study AG120-C-001, NCT02074839)of single-agent TIBSOVO®, for newly-diagnosed AML patients, with an IDH1 mutation detected by an FDA-approved IDH1 Assay. In this study, 28 patients were included and these patients were at least 75 years old, or had comorbidities that precluded the use of intensive induction chemotherapy. For comorbidities, enrolled patients met at least one of the following criteria – baseline ECOG PS of 2 or more, severe cardiac or pulmonary disease, hepatic impairment with Bilirubin more than 1.5 times the upper limit of normal, or Creatinine Clearance less than 45 mL/min. The median age was 77 years and 79% had therapy-related AML or AML with Myelodysplasia-related changes. Patients received TIBSOVO® 500 mg orally daily until disease progression, development of unacceptable toxicity, or hematopoietic stem cell transplantation. Efficacy was based on the rate of Complete Remission (CR) or Complete Remission with partial hematologic improvement (CRh) rate, the duration of CR+CRh, and the conversion rate from transfusion dependence to transfusion independence. CRh was defined as less than 5% of blasts in the bone marrow, no evidence of disease, and partial recovery of peripheral blood counts (platelets more than 50,000/microliter and ANC more than 500/microliter).
In this trial, TIBSOVO® demonstrated a CR+CRh rate of 42.9%, with a CR rate of 28.6% and a CRh rate was 14.3%. The median durations of CR and CR+CRh could not be estimated, with 41.7% of those who achieved CR or CRh remaining on TIBSOVO® treatment as of the data cutoff (treatment duration ranged from 20.3 to 40.9 months). At 12 months after receiving treatment, 58.3% of patients who achieved CR or CRh, remained in remission. For those who achieved a CR or CRh, the median time to best response of CR or CRh was 2.8 months. Among those patients who were dependent on RBC and/or platelet transfusions at baseline, 41.2% achieved transfusion independence lasting at least 8 weeks.
The most common adverse reactions were fatigue, nausea, diarrhea, rash, pyrexia, arthralgia, leukocytosis and QT prolongation. One important side effect of the IDH inhibitors is the induction of differentiation of the malignant cells, and in 10-20% of patients, a clinical syndrome known as the IDH differentiation syndrome can occur. The IDH differentiation syndrome should be promptly managed by dose interruption and treatment with glucocorticoids, oral hydroxyurea, or both.
It was concluded that TIBSOVO® can induce durable responses among newly diagnosed poor risk AML patients with an IDH1 mutation, who are ineligible for intensive chemotherapy, fulfilling an unmet need. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-ivosidenib-first-line-treatment-aml-idh1-mutation
TIBSOVO® (Ivosidenib)
The FDA on May 2, 2019 approved TIBSOVO® for newly diagnosed Acute Myeloid Leukemia (AML), with a susceptible IDH1 mutation, as detected by an FDA-approved test, in patients who are at least 75 years old or who have comorbidities that preclude the use of intensive induction chemotherapy. TIBSOVO® was approved in July 2018 for adult patients with relapsed or refractory AML with a susceptible IDH1 mutation. TIBSOVO® is a product of Agios Pharmaceuticals, Inc.
IMBRUVICA® plus GAZYVA® is a Safe and Effective Alternative First Line Treatment Option for CLL Patients with Comorbidities and High Risk Disease
SUMMARY: The American Cancer Society estimates that for 2019, about 20,720 new cases of Chronic Lymphocytic Leukemia (CLL) will be diagnosed in the US and 3,930 patients will die of the disease. B-cell CLL is the most common type of leukemia in adults, accounting for about 11% of all hematologic malignancies. The FDA in January 2019 approved IMBRUVICA® (Ibrutinib), a Bruton's Tyrosine Kinase Inhibitor, in combination with GAZYVA® (Obinutuzumab) for treatment-naive patients with CLL/Small Lymphocytic Lymphoma (CLL/SLL). This is the first approval of a non-chemotherapy combination regimen for treatment-naive patients with CLL/SLL.
Chronic Lymphocytic leukemia (CLL) is a disease of the elderly, with a median age at diagnosis of 71 years. Given the age at diagnosis, it is not uncommon for these patients to have multiple comorbidities. GAZYVA® is glycoengineered, fully humanized, third generation, type II anti-CD20 antibody (IgG1 monoclonal antibody) that selectivity binds to the extracellular domain of the CD20 antigen on malignant human B cells. By virtue of binding affinity of the glycoengineered Fc portion of GAZYVA® to Fcγ receptor III on innate immune effector cells such as natural killer cells, macrophages and neutrophils, Antibody-Dependent Cell-mediated Cytotoxicity (ADCC) and Antibody-Dependent Cellular phagocytosis is significantly enhanced, whereas it induces very little Complement-Dependent Cytotoxicity. This is in contrast to RITUXAN® (Rituximab), which is a first generation type I, chimeric anti-CD20 targeted monoclonal antibody that kills CLL cells primarily by Complement-Dependent Cytotoxicity and also ADCC. In a previously published study, the combination of GAZYVA® and LEUKERAN® (Chlorambucil) when given to elderly patients with comorbid conditions improved Overall Survival (OS) compared to LEUKERAN® alone, and resulted in higher Response Rates and longer Progression Free Survival (PFS) than RITUXAN® plus LEUKERAN® (NEJM 2014; 370:1101-1110).
IMBRUVICA® (Ibrutinib) is an oral, irreversible inhibitor of BTK and inhibits cell proliferation and promotes programmed cell death (Apoptosis) by blocking B-cell activation and signaling. IMBRUVICA® in phase III trials showed improved PFS and OS when compared to LEUKERAN® alone, in previously untreated, elderly patients with CLL (NEJM 2015; 373:2425-2437).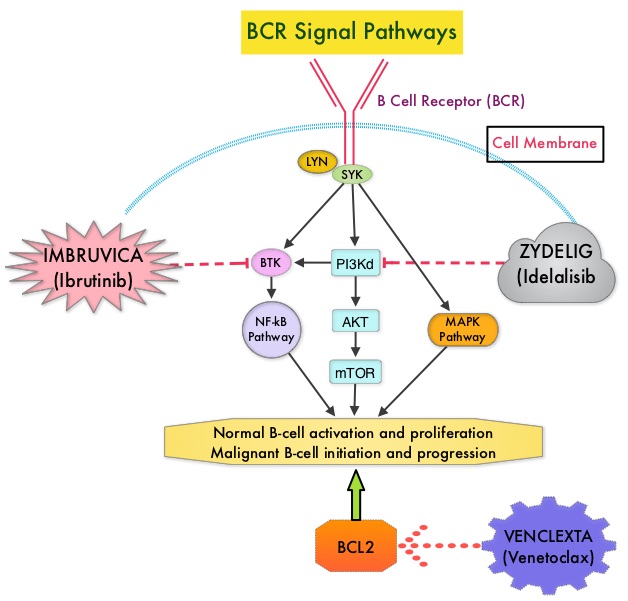
iLLUMINATE is a multicentre, randomized, open-label, international, Phase III trial which enrolled 229 patients with previously untreated CLL or Small Lymphocytic Lymphoma, either aged 65 years or older and if less than 65 years had at least one of the following criteria: Cumulative Illness Rating Scale (CIRS) more than 6, Estimated Creatinine Clearance of less than 70 mL/min using Cockcroft-Gault equation or del 17p by FISH or TP53 mutation by PCR or Next Generation Sequencing. (CIRS is a tool utilized to assess and quantify burden of comorbidity in individual patients). Patients were randomly assigned 1:1 to receive IMBRUVICA® plus GAZYVA® (N=113) or LEUKERAN® plus GAZYVA® (N=116). IMBRUVICA® plus GAZYVA® regimen consisted of IMBRUVICA® 420 mg PO once daily continuously combined with GAZYVA® 100 mg IV on day 1, 900 mg IV on day 2, 1000 mg IV on day 8, and 15 of cycle 1 and 1000 mg IV on day 1 of subsequent 28-day cycles, for a total of six cycles. LEUKERAN® plus GAZYVA® regimen consisted of LEUKERAN® 0.5 mg/kg PO on days 1 and 15 of each 28-day cycle for six cycles combined with GAZYVA® regimen as described above. Eighty percent (80%) of patients were 65 years or older and the median age was 71 years. Approximately 65% of patients had high-risk genetic abnormalities, 52% of patients had either Rai III or IV disease, with bulky disease at baseline in 27% of IMBRUVICA®-treated patients and 38% of LEUKERAN® treated patients. The Primary Endpoint was Progression Free Survival (PFS) and Secondary endpoints included PFS in High-risk Subpopulation which included those patients with del17p/TP53 mutation or del 11q deletion at baseline and/or unmutated IGHV disease. Patients who progressed on the LEUKERAN®treatment group were allowed by the IRC (Independent Review Committee) to cross over to the IMBRUVICA® treatment group.
At a median follow-up time was 31.3 months, the median PFS was significantly longer in the IMBRUVICA® plus GAZYVA® group compared to the LEUKERAN® plus GAZYVA® group ((median not reached versus 19.0 months (HR=0.23; P<0.0001), with a 77% reduction in the risk of progression or death. Patients with high-risk disease such as those with 17p deletion/TP53 mutation, 11q deletion, or unmutated immunoglobulin heavy chain variable region gene treated with IMBRUVICA® plus GAZYVA® experienced an 85% reduction in risk of progression or death (HR= 0.15). The IRC-evaluated Overall Response Rate was 89% in the IMBRUVICA® plus GAZYVA® group versus 73% in the LEUKERAN® plus GAZYVA® arm. The estimated 30-month PFS was 79% in the IMBRUVICA® plus GAZYVA® group and 31% in the LEUKERAN® plus GAZYVA® group. The most common Grade 3 or 4 adverse events in both treatment groups were neutropenia and thrombocytopenia.
It was concluded that a combination of IMBRUVICA® and GAZYVA® is a safe and effective chemotherapy-free regimen for previously untreated patients with CLL or Small Lymphocytic Lymphoma, independent of high-risk features, and provides an alternative first line treatment option for this patient group. Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): a multicentre, randomised, open-label, phase 3 trial. Moreno C, Greil R, Demirkan F, et al. Lancet Oncol. 2019;20:43-56.
KEYTRUDA® versus Chemotherapy as Second-Line Treatment for Advanced Esophageal Cancer
SUMMARY: The American Cancer Society estimates that in 2019, about 17,650 new cases of esophageal cancer will be diagnosed in the US and about 16,080 individuals will die of the disease. It is the sixth most common cause of global cancer death. Squamous Cell Carcinoma is the most common type of cancer of the esophagus among African Americans, while Adenocarcinoma is more common in caucasians. About 20% of patients survive at least 5 years following diagnosis. Patients with advanced esophageal cancer following progression on first line chemotherapy have limited treatment options and have a poor prognosis.
KEYTRUDA® (Pembrolizumab) is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2. It thereby reverses the PD-1 pathway-mediated inhibition of the immune response and unleashes the tumor-specific effector T cells. KEYTRUDA® in the Phase II KEYNOTE-180 study demonstrated durable responses among heavily pretreated patients with advanced metastatic Adenocarcinoma or Squamous Cell Carcinoma of the Esophagus as well as tumors with PD-L1 Combined Positive Score (CPS) of 10 or higher.
KEYNOTE-181 is a global, open-label, Phase III study which included 628 patients with advanced or metastatic adenocarcinoma or squamous cell carcinoma of the esophagus, or Siewert Type I adenocarcinoma of the esophagogastric junction that had progressed after first-line standard therapy. [Adenocarcinomas arising in the vicinity of the EsophagoGastric Junction are classified (Siewert classification) into adenocarcinoma of the distal esophagus (Type I), true carcinoma of the cardia (Type II) and subcardial carcinoma (Type III)].
Patients were randomized 1:1 to KEYTRUDA® 200 mg Q3W for up to 35 cycles (approximately2 years) or investigator’s choice chemotherapy with Docetaxel 75 mg/m2 IV on day 1 of each 21 day cycle, OR Paclitaxel 80-100 mg/m2 IV on days 1, 8 and 15 of each 28-day cycle, OR Irinotecan 80 mg/m2 IV on day 1 of each 14-day cycle. Randomization was stratified by histology and region (Asia vs rest of world). The majority of patients (N=401; 64%) had Squamous Cell Carcinoma (SCC), and 222 patients had PD-L1 Combined Positive Score (CPS) of 10 or higher. The three Primary end points were Overall Survival (OS) in patients with SCC, patients with PD-L1 CPS of 10 or higher and Intent-To- Treat populations. The median follow up was 7 months.
It was noted that among the patients with a PD-L1 CPS of 10 or higher (35% of the study population), the median Overall Survival was 9.3 months with KEYTRUDA® versus 6.7 months with chemotherapy (HR=0.69; P=0.0074). The 12-month survival rate in this group was 43% versus 20% respectively. In the Squamous Cell Carcinoma subgroup (N=401), the median Overall Survival was 8.2 months with KEYTRUDA® versus 7.1 months with chemotherapy (HR=0.78; P=0.0095). These differences favoring KEYTRUDA® however, did not meet the study’s prespecified statistical boundary. In the Intent-To- Treat population, the median Overall Survival was 7.1 months in each treatment group (HR=0.89; P=0.0560), and was not statistically significant. The Progression Free Survival at 12 months among patients with a PD-L1 CPS of 10 or higher was 21% versus 7% for KEYTRUDA® and chemotherapy, respectively. Further, in this patient group, KEYTRUDA® more than doubled the Response Rates than those achieved with chemotherapy, with a longer median duration of response (9.3 versus 7.7 months respectively). Fewer patients had any grade drug-related adverse events with KEYTRUDA®, compared with chemotherapy.
The authors concluded that KEYTRUDA® significantly improved Overall Survival compared with chemotherapy, as second line therapy for patients with advanced esophageal cancer, with PD-L1 CPS of 10 or higher and also had a more favorable safety profile. They added that these data support KEYTRUDA® as a new second line standard of care for esophageal cancer with PD-L1 CPS of 10 or higher. A Phase III study of KEYTRUDA® plus chemotherapy as first line therapy for advanced esophageal cancer is underway. Pembrolizumab versus chemotherapy as second-line therapy for advanced esophageal cancer: Phase III KEYNOTE-181 study. Kojima T, Muro K, Francois E, et al. J Clin Oncol 37, 2019 (suppl 4; abstr 2)