SUMMARY: ColoRectal Cancer (CRC) is the third most common cancer diagnosed in both men and women in the United States. The American Cancer Society estimates that approximately 145,600 new cases of CRC will be diagnosed in the United States in 2019 and about 51,020 patients are expected to die of the disease. The lifetime risk of developing CRC is about 1 in 23. Advanced colon cancer is often incurable and standard chemotherapy when combined with anti EGFR (Epidermal Growth Factor Receptor) targeted monoclonal antibodies such as VECTIBIX® (Panitumumab) and ERBITUX® (Cetuximab) as well as anti VEGF agent AVASTIN® (Bevacizumab), have demonstrated improvement in Progression Free Survival (PFS) and Overall Survival (OS). The benefit with anti EGFR agents however is only demonstrable in patients with metastatic CRC (mCRC), whose tumors do not harbor KRAS mutations in codons 12 and 13 of exon 2 (KRAS Wild Type). It is now also clear that even among the KRAS Wild Type patient group about 15-20% have other rare mutations such as NRAS and BRAF mutations, which confer resistance to anti EGFR agents. Patients with stage IV colorectal cancer are now routinely analyzed for extended RAS and BRAF mutations. KRAS mutations are predictive of resistance to EGFR targeted therapy. Approximately 8-15% of all metastatic CRC tumors present with BRAF V600E mutations and BRAF V600E is recognized as a marker of poor prognosis in this patient group. These patients tend to have aggressive disease with a higher rate of peritoneal metastasis and do not respond well to standard treatment intervention. Approximately 20% of the BRAF-mutated population in the metastatic setting has MSI-High tumors, but MSI-High status does not confer protection to this patient group.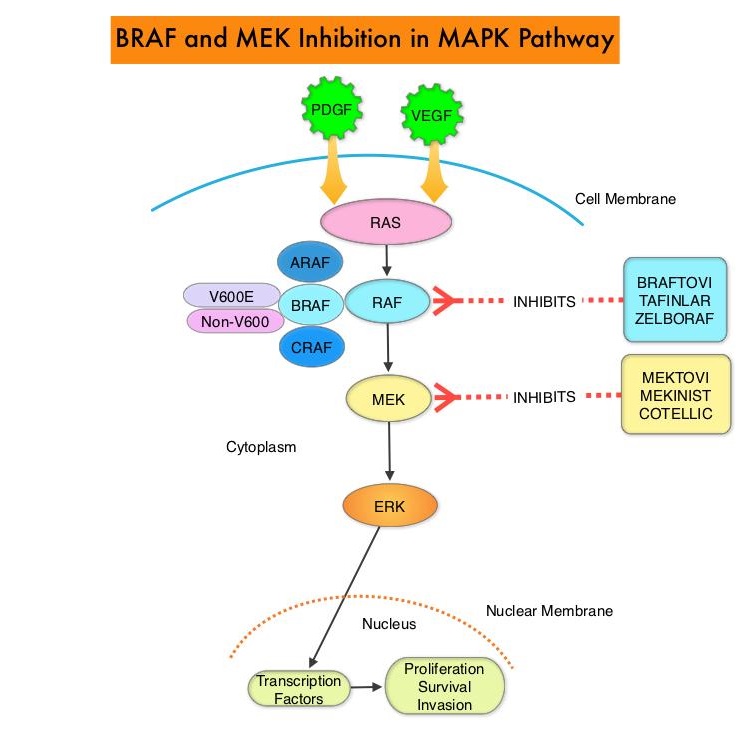
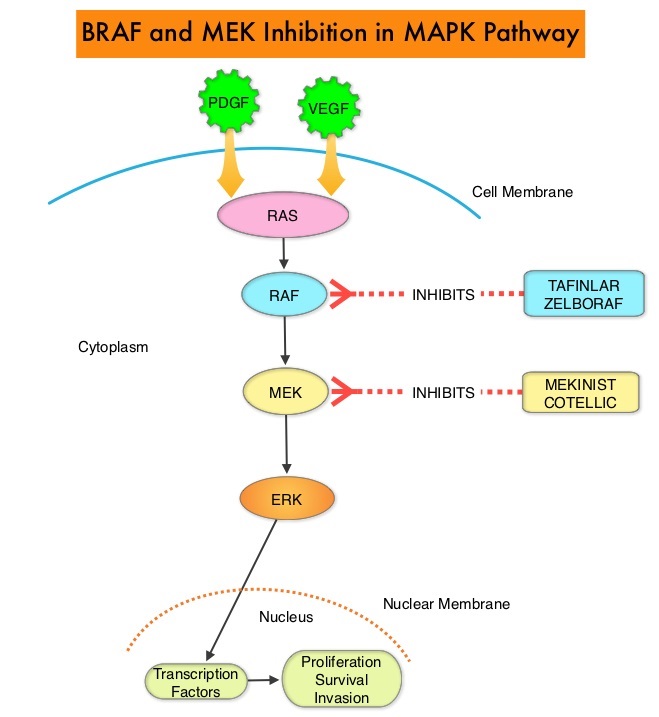
The Mitogen-Activated Protein Kinase pathway (MAPK pathway) is an important signaling pathway which enables the cell to respond to external stimuli. This pathway plays a dual role, regulating cytokine production and participating in cytokine dependent signaling cascade. The MAPK pathway of interest is the RAS-RAF-MEK-ERK pathway. The RAF family of kinases includes ARAF, BRAF and CRAF signaling molecules. BRAF is a very important intermediary of the RAS-RAF-MEK-ERK pathway. The BRAF V600E mutations results in constitutive activation of the MAP kinase pathway. Inhibiting BRAF can transiently reduce MAP kinase signaling. However, this can result in feedback upregulation of EGFR signaling pathway, which can then reactivate the MAP kinase pathway. This aberrant signaling can be blocked by dual inhibition of both BRAF and EGFR. However, BRAF V600E-mutated CRC is inherently less sensitive to BRAF inhibition than melanoma.
The FDA approved BRAFTOVI® (Encorafenib) and MEKTOVI® (Binimetinib) in combination for the first-line treatment of patients with BRAF V600-mutant melanoma, in June 2018. In a recent Phase II study among previously treated patients with BRAF V600E-mutant mCRC, treatment with a combination of BRAFTOVI® plus ERBITUX® resulted in an Objective Response Rate (ORR) of 24%, PFS of 4.2 months, and OS of 9.3 months. These results were significantly better than the standard of care, as well as other BRAF, MEK, and EGFR-inhibitor triplet combinations. Preclinical data suggests that BRAFTOVI® has target binding characteristics that differ from other BRAF inhibitors such as ZELBORAF® (Vemurafenib) and TAFINLAR® (Dabrafenib), with a prolonged target dissociation half-life and higher potency. This may explain the superior efficacy of BRAFTOVI® over other BRAF inhibitors in BRAF V600E-mutated CRC. These encouraging results with the BRAFTOVI® plus ERBITUX® doublet led to the initiation of the Phase III BEACON CRC study.
The BEACON Colorectal Cancer trial is an open-label, randomized, three-arm, Phase III study in which the efficacy and safety of BRAFTOVI® plus ERBITUX® with or without MEKTOVI® was compared with the investigators’ choice of ERBITUX® combined with either Irinotecan or Fluorouracil, Folinic acid, and Irinotecan, in patients with BRAF V600E-mutant mCRC whose disease has progressed after one or two prior regimens. At the time BEACON CRC was initiated, the triplet combination of MEKTOVI®, BRAFTOVI®, and ERBITUX® had not been clinically evaluated. The authors therefore conducted a Safety Lead-In (SLI) to determine the Safety, tolerability, and preliminary efficacy of this triplet combination at the same doses planned for the randomized portion of the trial.
The randomized portion of the trial was ongoing at the time of this analysis. The BEACON trial included patients with mCRC whose tumor tissue was positive for the presence of BRAF V600E mutation. Majority of the patients had right-sided disease as is characteristic of BRAF V600E-mutant mCRC, with a high frequency of nodal and peritoneal metastasis. Liver however, was the most frequent site of metastasis. Enrolled patients must have had progressive disease on one, but no more than two prior treatment regimens, in the metastatic setting. Enrolled patients received BRAFTOVI® 300 mg PO daily plus MEKTOVI® 45 mg PO BID along with ERBITUX® 400 mg/m2 IV, followed by 250 mg/m2 IV weekly every 28 days. The Safety Lead-In was initiated before the randomized portion of the BEACON trial and included 30 patients with disease characteristics and treatment schedule as described above. The Primary end point was Safety, including the incidence of dose-limiting toxicities. Efficacy end points included Overall Response Rate, Progression Free Survival, and Overall Survival.
The median follow up was 18.2 months, and the median time on study drug was 7.9 months. The confirmed Overall Response Rate was 48%, median Duration of Response was 5.5 months, median Progression Free Survival was 8.0 months, and median Overall Survival was 15.3 months. Approximately 79% of responding patients achieved a response within 2 months. The most common adverse events were nausea, diarrhea, fatigue and dermatitis. Approximately 6% of patients experienced serous retinopathy without loss of visual acuity.
It was concluded that in this Safety Lead-In, the combination regimen of BRAFTOVI®, MEKTOVI® and ERBITUX® resulted in promising results compared with available therapies, among patients with previously treated BRAF V600E-mutant mCRC, and if confirmed in the randomized portion of the trial, could become the new standard of care in this patient group. Binimetinib, Encorafenib, and Cetuximab Triplet Therapy for Patients With BRAF V600E–Mutant Metastatic Colorectal Cancer: Safety Lead-In Results From the Phase III BEACON Colorectal Cancer Study. Van Cutsem E, Huijberts S, Grothey A, et al. Journal of Clinical Oncology 2019; 37:1460-1469.