
The FDA on December 20, 2019 granted accelerated approval to ENHERTU® for patients with unresectable or metastatic HER2-positive breast cancer, who have received two or more prior anti-HER2-based regimens in the metastatic setting. ENHERTU® is a product of Daiichi Sankyo.
Month: December 2019
PADCEV® (Enfortumab vedotin-ejfv)
The FDA on December 18, 2019 granted accelerated approval to PADCEV® for adult patients with locally advanced or metastatic urothelial cancer who have previously received a Programmed Death receptor-1 (PD-1) or Programmed Death-Ligand 1 (PD-L1) inhibitor, and a Platinum-containing chemotherapy in the neoadjuvant/adjuvant, locally advanced or metastatic setting. PADCEV® is a product of Astellas Pharma US, Inc.
XTANDI® (Enzalutamide)
The FDA on December 16, 2019 approved XTANDI® for patients with metastatic Castration-Sensitive Prostate Cancer (mCSPC). XTANDI® is a product of Astellas Pharma Inc.
TECENTRIQ® (Atezolizumab)
The FDA on December 3, 2019 approved TECENTRIQ® in combination with Paclitaxel protein-bound and Carboplatin for the first-line treatment of adult patients with metastatic non-squamous Non-Small Cell Lung Cancer (NSCLC) with no EGFR or ALK genomic tumor aberrations. TECENTRIQ® is a product of Genentech Inc.
FDA Approves ENHERTU® for Advanced HER2-Positive Breast Cancer
SUMMARY: The FDA on December 20, 2019, granted accelerated approval to ENHERTU® (Trastuzumab deruxtecan) for patients with unresectable or metastatic HER2-positive breast cancer, who have received two or more prior anti-HER2-based regimens in the metastatic setting. Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 268,600 new cases of invasive breast cancer will be diagnosed in 2019 and about 41,760 women will die of the disease. The HER or erbB family of receptors consist of HER1, HER2, HER3 and HER4. Approximately 15-20% of invasive breast cancers overexpress HER2/neu oncogene, which is a negative predictor of outcomes without systemic therapy. Patients with HER2-positive metastatic breast cancer are often treated with anti-HER2 targeted therapy along with chemotherapy, irrespective of hormone receptor status, and this has resulted in significantly improved treatment outcomes. HER2-targeted therapies include HERCEPTIN® (Trastuzumab), TYKERB® (Lapatinib), PERJETA® (Pertuzumab) and KADCYLA® (ado-Trastuzumab emtansine). Dual HER2 blockade with HERCEPTIN® and PERJETA®, given along with chemotherapy (with or without endocrine therapy), as first line treatment, in HER2 positive metastatic breast cancer patients, was shown to significantly improve Progression Free Survival (PFS) as well as Overall Survival (OS). The superior benefit with dual HER2 blockade has been attributed to differing mechanisms of action and synergistic interaction between HER2 targeted therapies. Patients progressing on Dual HER2 blockade often receive KADCYLA® which results in an Objective Response Rate (ORR) of 44% and a median PFS of 9.6 months, when administered after HERCEPTIN® and a taxane. There is however no standard treatment option for this patient population following progression on KADCYLA®.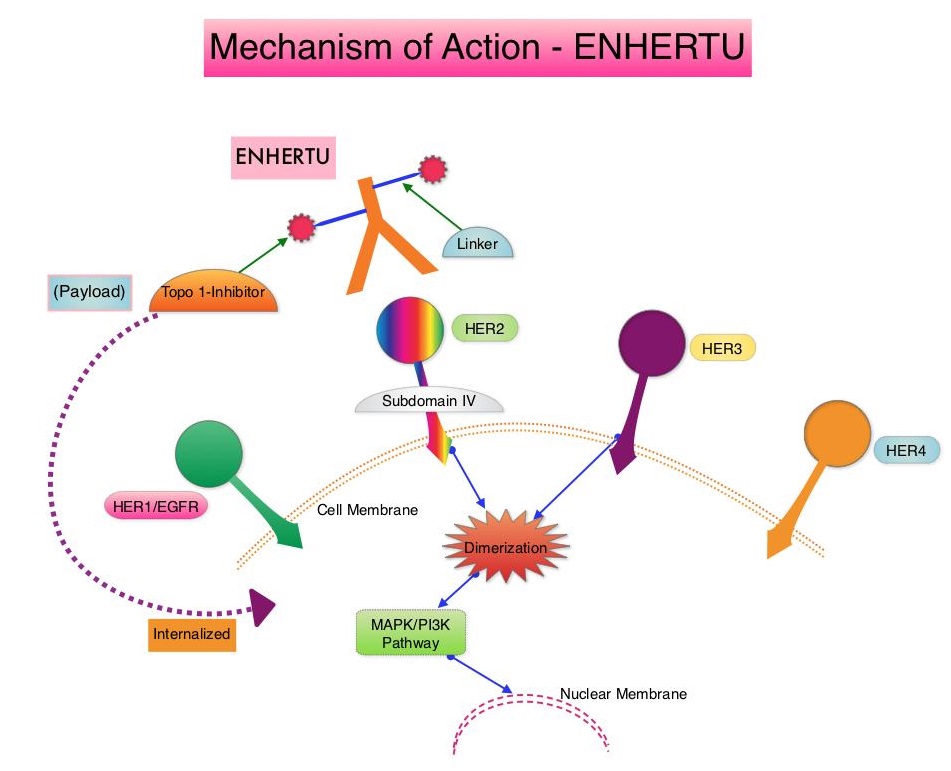
ENHERTU® is an Antibody-Drug Conjugate (ADC) composed of a humanized monoclonal antibody specifically targeting HER2, with the amino acid sequence similar to Trastuzumab, a cleavable tetrapeptide-based linker, and a potent cytotoxic Topoisomerase I inhibitor as the cytotoxic drug (payload). ENHERTU® has a favorable pharmacokinetic profile and the tetrapeptide-based linker is stable in the plasma and is selectively cleaved by cathepsins that are up-regulated in tumor cells. Unlike KADCYLA®, ENHERTU® has a higher drug-to-antibody ratio (8 versus 4), released payload easily crosses the cell membrane with resulting potent cytotoxic effect on neighboring tumor cells regardless of target expression, and the released cytotoxic agent (payload) has a short half-life, minimizing systemic exposure. In a Phase 1 dose-finding study involving patients with advanced HER2-positive breast cancer, treatment with ENHERTU® resulted in a confirmed response rate was 59.5%, and the median response duration was 20.7 months. However, the efficacy of ENHERTU® in patients with HER2-positive metastatic breast cancer, previously treated with KADCYLA® remained unclear.
The present FDA approval was based on DESTINY-Breast01 study, which is a multicenter, single-arm, Phase II trial, in which 184 patients with HER2-positive, metastatic breast cancer, who had received two or more prior HER2 targeted therapies including KADCYLA®, were enrolled. Patients received ENHERTU® 5.4 mg/kg IV every 3 weeks until disease progression or unacceptable toxicity. The median age was 55 years, 53% had Hormone Receptor-positive tumors and the median number of previous lines of therapy for metastatic disease was six and included KADCYLA® (100%), Trastuzumab (100%), Pertuzumab (66%), and other anti-HER2 therapies (54%). The Primary end point was Objective Response Rate (ORR) assessed by Independent Central Review and Secondary endpoints included Duration of Response, Progression Free Survival (PFS) and Overall Survival (OS). The median follow up was 11.1 months. The ORR was 60.9%, with 6% Complete Responses and 54.9% Partial Responses. The median time to response was 1.6 months and the median response duration was 14.8 months. The median PFS was 16.4 months the median OS was not reached at the time of this publication. The most Grade 3 or higher adverse events were cytopenias, nausea, diarrhea and Interstitial Lung Disease.
It was concluded that ENHERTU® has a high level of clinical efficacy with a durable antitumor activity in a pretreated patient population with HER2-positive metastatic breast cancer. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. Modi S, Saura C, Yamashita T, et al. for the DESTINY-Breast01 Investigators. N Engl J Med. 2019 Dec 11. doi: 10.1056/NEJMoa1914510. [Epub ahead of print]
FDA Grants Accelerated Approval to PADCEV® for Metastatic Urothelial Cancer
SUMMARY: The FDA on December 18, 2019, granted accelerated approval to PADCEV® (Enfortumab vedotin-ejfv), for adult patients with locally advanced or metastatic urothelial cancer who have previously received a Programmed Death receptor-1 (PD-1) or Programmed Death-Ligand1 (PD-L1) inhibitor, and a Platinum-containing chemotherapy in the neoadjuvant/adjuvant, locally advanced or metastatic setting. The American Cancer Society estimates that in 2019, approximately 80,470 new cases of Bladder Cancer will be diagnosed and 17,670 patients will die of the disease. Patients with urothelial carcinoma are currently treated in the first line setting with a Platinum based chemotherapy regimen and a Check Point Inhibitor (PD-1 or PD-L1 inhibitor) in the second line setting. Treatment options for patients who progress after first and second line therapies are limited, with poor outcomes. The response rates with standard chemotherapy in this patient population, is about 10%.
PADCEV® is an Antibody-Drug Conjugate (ADC) that targets Nectin-4, a cell adhesion molecule highly expressed in urothelial cancers and other solid tumors. Following binding to Nectin-4 on the cell surface, PADCEV® becomes internalized and is processed by lysosomes, with the liberation of its cytotoxic payload, Monomethyl auristatin E, which in turn disrupts microtubule assembly, leading to cell cycle arrest and apoptosis. In a Phase I dose-finding study of PADCEV®, the Objective Response Rate (ORR) was 42% among patients with advanced urothelial cancer, who previously received treatment with a PD-1/PD-L1 inhibitor.
This FDA approval was based on the results from the pivotal Phase II EV-201 study, which is an open-label, single-arm, multicenter trial in which 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and Platinum-based chemotherapy were enrolled. Patients received PADCEV® 1.25 mg/kg on days 1, 8, and 15 of a 28-day cycle, until disease progression or unacceptable toxicity. The median age was 69 years. The Primary endpoint was ORR as assessed by blinded Independent Central Review. Secondary endpoints included Duration of Response, Progression Free Survival (PFS), Overall Survival (OS), Safety and Tolerability.
The ORR was 44%, with 12% Complete Responses and 32% Partial Responses. Overall, 84% of evaluable patients showed some degree of tumor shrinkage. The responses were noted at a median of 1.8 months after treatment initiation and the median Duration of Response was 7.6 months. These objective responses were seen in all patient subgroups evaluated, including those with poor prognostic features. The median PFS was 5.8 months, and the median Overall Survival was 11.7 months. The most common adverse reactions were fatigue, alopecia, decreased appetite and peripheral neuropathy. Blood glucose levels should be monitored closely in patients with, or at risk, for diabetes mellitus or hyperglycemia.
It was concluded from this study that treatment with PADCEV® demonstrated clinically meaningful Objective Response Rate, in patients with advanced metastatic urothelial cancer, who received prior treatment with a PD-1 or PD-L1 inhibitor and Platinum-based chemotherapy, thus fulfilling an unmet need. PADCEV® is the first Nectin-4-directed Antibody-Drug Conjugate to receive FDA approval, and a Phase III study is underway comparing PADCEV® against standard single-agent chemotherapy, in patients with advanced, previously treated metastatic urothelial cancer. EV-201: Results of enfortumab vedotin monotherapy for locally advanced or metastatic urothelial cancer previously treated with platinum and immune checkpoint inhibitors. Petrylak DP, Balar AV, O’Donnell PH, et al. DOI: 10.1200/JCO.2019.37.18_suppl.LBA4505 Journal of Clinical Oncology 37, no. 18_suppl (June 20, 2019) 4505-4505.
DARZALEX® Combination Improves Overall Survival in Transplant-Ineligible Myeloma Patients
SUMMARY: Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, 32,110 new cases will be diagnosed in 2019 and 12,960 patients are expected to die of the disease. Multiple Myeloma (MM) in 2019 remains an incurable disease. The therapeutic goal therefore is to improve Progression Free Survival (PFS) and Overall Survival (OS). Multiple Myeloma is a disease of the elderly, with a median age at diagnosis of 69 years and characterized by intrinsic clonal heterogeneity. Almost all patients eventually will relapse, and patients with a high-risk cytogenetic profile or refractory disease have the worst outcomes. The median survival for patients with myeloma is over 10 years.
Elderly patients with myeloma in the US are often treated with a combination of REVLIMID® (Lenalidomide) and Dexamethasone, whereas Melphalan, Prednisone, and Thalidomide (MPT) and VELCADE® (Bortezomib), Melphalan and Prednisone (VMP) are the most widely used regimens outside the US. These regimens are associated with a PFS of 18-24 months and an OS of 4-5 years. For patients with newly diagnosed multiple myeloma who are ineligible for ASCT, treatment with VMP regimen has been a standard effective regimen, based on the VISTA (Velcade as Initial Standard Therapy in Multiple Myeloma: Assessment with Melphalan and Prednisone) trial.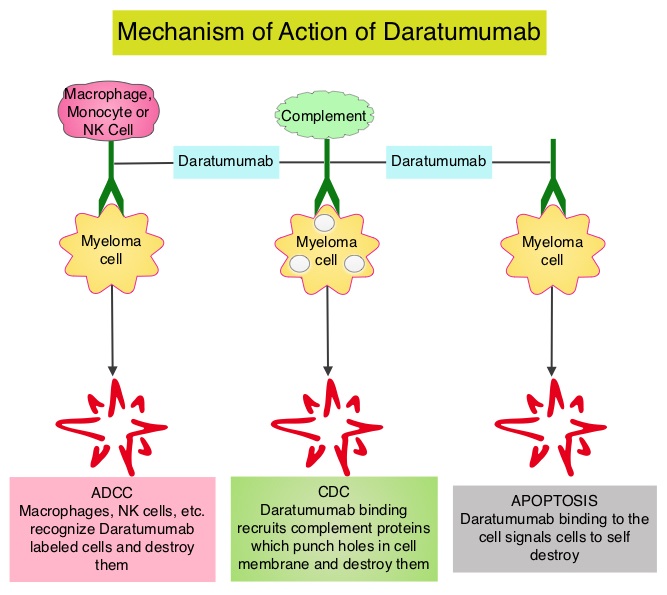
DARZALEX® (Daratumumab) is a human IgG1 antibody that targets CD38, a transmembrane glycoprotein abundantly expressed on malignant plasma cells and with low levels of expression on normal lymphoid and myeloid cells. DARZALEX® exerts its cytotoxic effect on myeloma cells by multiple mechanisms, including Antibody Dependent Cellular Cytotoxicity (ADCC), Complement Dependent Cytotoxicity (CDC) and direct Apoptosis. Additionally, DARZALEX® may have a role in immunomodulation, by depleting CD38-positive regulator immune suppressor cells, and thereby expanding T cells, in patients responding to therapy.
ALCYONE is a multicenter, randomized, open-label, active-controlled, Phase III trial in which DARZALEX® given along with VELCADE®, Melphalan and Prednisone (D-VMP regimen) was compared with VMP alone (control group), in patients with newly diagnosed multiple myeloma, who were ineligible for Autologous Stem Cell Transplantation (ASCT). Of the 706 enrolled patients, 350 were assigned to the DARZALEX® group and 356 to the control group. The median age was 71 yrs. All the patients received up to nine 6 week cycles of VELCADE® 1.3 mg/m2 SQ, twice weekly on weeks 1, 2, 4, and 5 of cycle 1 and once weekly on weeks 1, 2, 4, and 5 of cycles 2-9), Melphalan 9 mg/m2 orally, once daily on days 1-4 of each cycle, and Prednisone 60 mg/m2 once daily on days 1-4 of each cycle. In the study group, patients received DARZALEX® 16 mg/kg IV administered with Dexamethasone 20 mg oral or IV (to manage infusion reactions), once weekly for a total of 6 doses, every 3 weeks for a total of 16 doses and every 4 weeks thereafter until disease progression or unacceptable toxicity. The Primary end point was Progression Free Survival (PFS). Secondary end points included Overall Response Rate (ORR), rates of Very Good Partial Response (VGPR), Complete Response (CR) rate, Minimal Residual Disease (MRD) negativity and Overall Survival (OS). The FDA in 2018 approved DARZALEX® in combination with VELCADE® (Bortezomib), a proteasome inhibitor, Melphalan, an alkylating agent and Prednisone VMP regimen), for the treatment of patients with newly diagnosed multiple myeloma who are ineligible for Autologous Stem Cell Transplant (ASCT), based on the Progression Free Survival (PFS) benefit at 16.5 months, noted during the primary analysis of the ALCYONE study. The authors herein presented outcomes after more than 36 months of follow-up from the ALCYONE study, including analysis of Overall Survival (OS) from a prespecified interim analysis.
In this updated analysis, treatment with D-VMP continued to demonstrate a twofold greater median PFS at 36.4 months versus 19.3 months with VMP, after a median follow-up of 41months (HR=0.42; P<0.0001). Patients assigned to D-VMP also had significantly prolonged PFS on subsequent therapy (PFS-2). Median PFS-2 was not reached with D-VMP versus 42.3 months with VMP (HR=0.55; P<0.0001), representing a 45% reduction in the risk for progression or death. The median time to subsequent therapy had yet to be reached in the D-VMP group versus 25.9 months for the VMP group. The Overall Response Rate was 91% in the D-VMP group as compared with 74% in the control group and the rate of Complete Response or better (including stringent CR) was 46%, versus 24.4% in the control group. The MRD-negative rate (at a threshold of 1 tumor cell per 105 white cells) was 28% with D-VMP and 7% with VMP and D-VMP also led to higher rates of sustained MRD negativity. MRD negativity for 12 or more months was associated with improved PFS. The estimated 36 month OS rate was 78% with D-VMP versus 68% with VMP, with a significant benefit for OS observed for D-VMP versus VMP alone (HR=0.60; P=0.0003). This represented a 40% reduction in the risk of death, in favor of D-VMP.
The authors concluded that for the first time, this study demonstrated that the addition of DARZALEX® to VMP significantly prolonged Overall Survival in patients with transplant-ineligible newly diagnosed Multiple Myeloma, with a 40% reduction in the risk of death, when compared with VMP alone. They added that these findings, together with the Phase 3 MAIA study (DARZALEX® plus Lenalidomide and Dexamethasone versus Lenalidomide plus Dexamethasone), continue to support the addition of DARZALEX® to frontline treatment regimens for patients with Multiple Myeloma. Daratumumab Plus Bortezomib, Melphalan, and Prednisone Versus Bortezomib, Melphalan, and Prednisone in Patients with Transplant-Ineligible Newly Diagnosed Multiple Myeloma: Overall Survival in Alcyone. Mateos MV, Cavo M, Bladé J, et al. Presented at: 2019 ASH Annual Meeting; December 7-10, 2019; Orlando, FL. Abstract 859.
Long-Term Breast Cancer Preventive Benefit with ARIMIDEX®
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 268,600 new cases of invasive breast cancer will be diagnosed in 2019 and about 41,760 women will die of the disease. Some high risk factors for the development of breast cancer include first-degree relative with breast cancer at any age, first-degree relative with bilateral breast cancer who developed the first breast cancer at 50 years of age or less, Lobular Carcinoma In Situ (LCIS), Atypical ductal or lobular hyperplasia in a benign lesion, and Ductal Carcinoma In-Situ (DCIS).
Previously published studies have shown a 38% reduction in all breast cancers and 50% reduction of ER-positive tumors with the use of SERMs (Selective Estrogen Receptor Modulators) such as Tamoxifen and Raloxifene for breast cancer prevention. Further it has been shown that the effects of Tamoxifen continue with a constant 29% annual preventive effect for at least 15 years after completion of treatment. A further improvement in breast cancer incidence short-term was seen in two trials using, two Aromatase Inhibitors, ARIMIDEX® (Anastrozole) in the International Breast Cancer Intervention Study II (IBIS-II) and AROMASIN® (Exemestane) in the MAP.3 trial. However in the MAP.3 study, the study was unblinded after the initial publication and a post-treatment effect, as was seen with Tamoxifen, was not possible.
The International Breast Cancer Intervention Study II (IBIS-II) is an international, randomized, double-blind, placebo-controlled trial, which was initiated in 2003. In this trial, 3864 postmenopausal women aged 40-70 years, at increased risk of developing breast cancer were recruited and were randomly assigned (1:1) to either ARIMIDEX® 1 mg orally daily (N=1920) or matching placebo (N=1944) daily for 5 years. After treatment completion, women were followed on a yearly basis and data was collected on breast cancer incidence, death, incidence of other cancers, and major adverse events (cardiovascular events and fractures). The median age at study entry was 59 years. The exclusion criteria for this study included premenopausal status, prior breast cancer including Ductal Carcinoma In Situ (DCIS) diagnosed more than 6 months before trial entry, current or previous Tamoxifen, Raloxifene, or other SERM use for more than 6 months, or previous or planned prophylactic mastectomy. Unblinding was only permitted if the participant developed breast cancer, when a clinician considered there to be valid medical or safety reasons. The Primary outcome was the development of histologically confirmed breast cancer, either invasive or non-invasive (DCIS), particularly during the post-5-year time period. Secondary outcomes were ER-positive breast cancer, breast cancer mortality, incidence of other cancers, cardiovascular disease, fractures, and all-cause mortality. The decision to analyze the data was made without looking at the results before hand. The first analysis after a median follow-up of 60 months showed a significant reduction (53%) in incidence for all breast cancer (including DCIS). The authors now report the results on the extended duration of benefit of ARIMIDEX® in preventing breast cancer, for up to 12 years after trial entry.
After a median follow up of 10.9 years for this analysis, women assigned to the ARIMIDEX® group were 49% less likely to develop breast cancer than women assigned to the placebo arm of the study ((HR=0.51, P<0.0001). The reduction in incidence in the first 5 years of follow up was 61% (HR=0.39; P<0.0001), and a smaller but still significant 36% reduction (HR=0.64; P=0•014) was seen in subsequent years, which was still larger than that seen for Tamoxifen in previous trials, and the effects in the two time periods was not significantly different (P=0.08). Invasive ER-positive breast cancer was reduced by 54% with ARIMIDEX® treatment (HR=0.46; P<0.0001), with a continued significant effect observed in the post-treatment follow up period. A 59% reduction in DCIS overall was observed (HR=0.41; P=0.0081), with a very large reduction noted in those cases known to be ER-positive (HR = 0.22; P<0.0001). A significant decrease in non-breast cancers was observed in the ARIMIDEX® group, primarily contributed by non-melanoma skin cancer (P=0.0042), and no excess rates of fractures or cardiovascular disease was observed.
The authors concluded that this updated analysis shows a continuing long-term effect of 5 years of ARIMIDEX® treatment, in preventing breast cancer, in high-risk postmenopausal women. These new results strongly suggest that ARIMIDEX® should be preferred therapy for breast cancer prevention in postmenopausal women at increased risk for the disease, with Tamoxifen used for women who experience severe side effects from ARIMIDEX®. Use of anastrozole for breast cancer prevention (IBIS-II): long-term results of a randomised controlled trial. Cuzick J, Sestak I, Forbes JF, et al. The Lancet. Published:December 12, 2019. DOI:https://doi.org/10.1016/S0140-6736(19)32955-1
Supplemental MRI Screening for Women with Extremely Dense Breast Tissue
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 268,600 new cases of invasive breast cancer will be diagnosed in 2019 and about 41,760 women will die of the disease. Screening mammography complemented by breast self exam and clinical breast exam has resulted in early detection of breast cancer and successful outcomes. Even though mammography is a sensitive screening test, a small percentage of breast cancers may not show up on mammograms but may be palpable on examination by the patient or the clinician. Further, mammograms are less likely to find breast tumors in younger women with dense breast tissue. A breast Magnetic Resonance Imaging (MRI) is more sensitive than mammography although the specificity of a breast MRI is lower, resulting in a higher rate of false-positive findings and potentially unnecessary biopsies. Microcalcifications in the breast can be missed by a breast MRI. Taking these factors into consideration, appropriate utilization of breast MRI becomes relevant.
The American Cancer Society (ACS) recommends an annual MRI as an adjunct to screening mammogram and clinical breast exam in certain groups with increased risk of breast cancer. In a study published by Stout NK, et al., (JAMA Intern Med. 2014;174:114-121), it was noted that breast MRI was over utilized in those who did not fit the ACS criteria and was under utilized in those with documented genetic mutations. Routine breast MRI screening is not recommended for a new breast cancer diagnosis or for breast cancer surveillance and should only be considered for the group of individuals who have the most benefit. Breast MRI is performed preferably between days 7-15 of menstrual cycle for premenopausal women, using a dedicated breast coil, with the ability to perform a biopsy under MRI guidance by experienced radiologists, during the same visit.
DENSE trial is a multicenter, randomized, controlled study which evaluated the effect of supplemental Magnetic Resonance Imaging (MRI) on the incidence of interval cancers, in women with extremely dense breast tissue. In this trial, 40,373 women between the ages of 50 and 75 years with extremely dense breast tissue and normal results on screening mammography were randomly assigned in a 1:4 ratio to a group that was invited to undergo supplemental MRI (N=8061) or to a group that received mammography screening alone (N=32,312). Of the women who were invited to undergo MRI, 59% accepted the invitation (N=4783). All MRI examinations were performed with the use of a dedicated bilateral breast coil. The Primary endpoint was the difference in the incidence of interval cancers during a 2-year screening period, between the mammography screening-only group and MRI-invitation group. Secondary endpoints included the recall rate for additional examination, the cancer-detection rate on MRI, the false positive rate, the positive predictive value, and tumor characteristics.
The interval cancer rate was 2.5 per 1000 screenings in the MRI-invitation group and 5.0 per 1000 screenings in the mammography-only group (P<0.001). The MRI cancer-detection rate among the women who actually underwent MRI screening was 16.5 per 1000 screenings. The Positive Predictive Value of a positive MRI result was 17.4%, the Positive Predictive Value of an indication for biopsy was 23.9% and the Positive Predictive Value of a biopsy was 26.3%. The false positive rate was 79.8 per 1000 screenings. As a result of the MRI screening, 300 women underwent a breast biopsy and of these women, breast cancer was diagnosed in 79 women, of whom 64 had invasive breast cancer and 15 were diagnosed with DCIS.
The authors concluded that the use of supplemental MRI screening in women with extremely dense breast tissue and normal results on mammography resulted in the diagnosis of significantly fewer interval cancers than mammography alone, during a 2-year screening period. Whether a reduction in interval cancers is an appropriate surrogate for improved Overall Survival, remains unclear. Supplemental MRI Screening for Women with Extremely Dense Breast Tissue. Bakker MF, de Lange SV, Pijnappel RM, et al. for the DENSE Trial Study Group. N Engl J Med 2019; 381:2091-2102
FDA Approves Frontline TECENTRIQ® with Carboplatin and nab-Paclitaxel for Metastatic Non-Squamous NSCLC
SUMMARY: The FDA on December 3, 2019 approved TECENTRIQ® (Atezolizumab) in combination with nab-Paclitaxel and Carboplatin for the first-line treatment of adult patients with metastatic non-squamous Non-Small Cell Lung Cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations. Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2019, about 228,150 new cases of lung cancer will be diagnosed and 142,670 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions by switching off the T cells of the immune system. Immuno-Oncology (IO) therapies unleash the T cells by blocking the Immune checkpoint proteins, thereby resulting in T cell proliferation, activation and a therapeutic response. Immunotherapy with PD-1 (Programmed cell Death 1) and PD-L1 (Programmed cell Death Ligand 1) inhibitors have demonstrated a clear survival benefit both as a single agent or in combination, compared with standard chemotherapy, in both treatment-naive and previously treated patients for advanced NSCLC. It is now standard therapy for patients with lung cancer. TECENTRIQ® is an anti PD-L1 monoclonal antibody, designed to directly bind to PD-L1, expressed on tumor cells and tumor-infiltrating immune cells, thereby blocking its interactions with PD-1 and B7.1 receptors and thus enabling the activation of T cells. 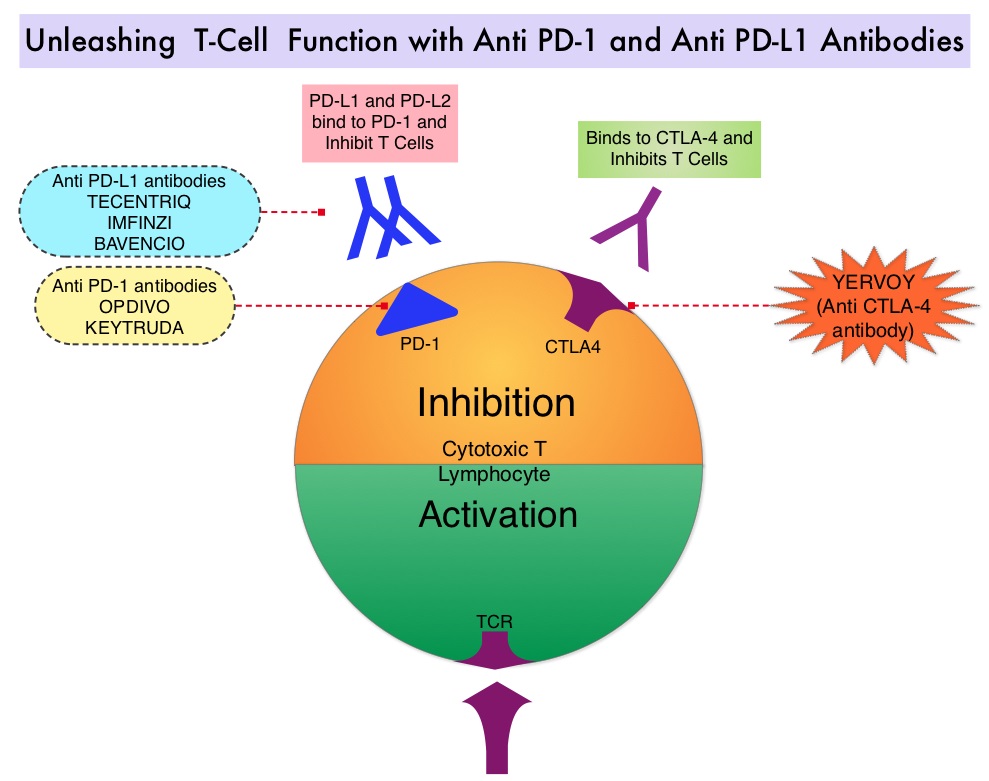
IMpower 130 is an international, multicentre, open-label, randomized, Phase III study, evaluating the efficacy and safety of TECENTRIQ® in combination with Carboplatin and nab-Paclitaxel versus chemotherapy (Carboplatin and nab-Paclitaxel) alone, for chemotherapy-naïve patients with Stage IV non-squamous NSCLC. This study enrolled 724 patients who were randomly assigned 2:1 to receive TECENTRIQ® 1200 mg IV on Day 1, along with Carboplatin AUC 6 on Day 1 and nab-Paclitaxel 100 mg/m2 IV, on days 1, 8 and 15 of each 21-day cycle, for 4 or 6 cycles followed by maintenance TECENTRIQ®, or Carboplatin and nab-Paclitaxel alone (control group), followed by Best Supportive Care during the maintenance treatment phase or Switch maintenance to Pemetrexed every 3 weeks. Stratification factors included gender, baseline liver metastases, and PD-L1 expression. The co-Primary endpoints were investigator-assessed Progression Free Survival and Overall Survival in the intention-to-treat EGFR and ALK wild-type population. The median follow up in the population studied was 19 months.
There was a significant improvement in median Overall Survival in the TECENTRIQ® plus chemotherapy group at 18.6 months compared to 13.9 months in the chemotherapy alone group (HR=0.79; P=0.033), as well as improvement in the median Progression Free Survival (7 months versus 5.5 months, respectively, HR=0.64; P<0.0001). The most common adverse reactions reported in 20% or more of patients in the TECENTRIQ® and chemotherapy group were fatigue/asthenia, nausea, alopecia, constipation, diarrhea, and decreased appetite.
It was concluded that first-line TECENTRIQ® in combination with chemotherapy significantly improved Overall Survival and Progression Free Survival, compared to chemotherapy alone, in patients with advanced non-squamous NSCLC without ALK or EGFR mutations. This IO-chemotherapy combination is the second FDA approval for this patient population. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): a multicentre, randomised, open-label, phase 3 trial. West H, McCleod M, Hussein M, et al. Lancet Oncol. 2019;20:924-937